

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Familial benign hypocalciuric hypercalcemia (FBHH) is a rare autosomal dominant disorder exhibiting benign hypercalcemia, inappropriately normal parathyroid hormone (PTH) levels and relative hypocalciuria, thus reflecting partial resistance to the normal effects of extracellular calcium on parathyroid glands and kidneys.1 The CaSR is expressed abundantly in the parathyroid gland, thyroid C cells, and kidney. The vast majority of FBHH kindred's have their genetic abnormality link to chromosome 3q21-24, the locus for the CaSR gene.2,3 We report a Korean family with FBHH carrying a mutation in the CaSR gene.
CASE REPORT
A 38-year-old male admitted to the hospital because of a known 3 cm sized right renal mass detected by abdominal ultrasonography in routine health check up and confirmed by computed tomography. He underwent right radical nephrectomy and the pathologic report revealed clear cell type renal cell carcinoma with capsular invasion, but without distant metastasis. His serum calcium level was 10.9 mg/dL (normal, 8.8-10.8) at preoperative evaluation but was ignored. One year after surgery for a follow-up visit, he was referred to the Endocrinology and Metabolism department due to persistent hypercalcemia. The serum calcium level was 11.1 mg/dL and the inorganic phosphorus was 3.5 mg/dL (normal, 2.5-4.5). The serum alkaline phosphatase level was 67 U/L (normal, 30-120), intact PTH was 32 pg/mL (normal, 10-65), and urine calcium was 20 mg/day (normal, 100-300). A ratio of urine calcium creatinine clearance was 0.0019. Thyroid and adrenal hormone levels were normal. There was no evidence of local recurrence or metastasis of carcinoma. The bone mineral density of the femoral neck and lumbar spines performed by dual energy x-ray absorptiometry (Expert XL, Lunar Co., Madison, WI, USA) showed normal (femoral neck T-score, 1.0; lumbar spines T-score, -0.6). The biochemical and clinical evaluation was consistent with FBHH.
Genomic DNA was isolated from whole blood samples using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) after informed consent was obtained. DNA sequence of CaSR gene of the proband was analyzed by PCR amplification of all coding exons of CaSR gene and direct sequencing of PCR products. Briefly, the PCR amplification was carried out in a PTC-225 DNA Engine Tetrad (MJ Research, Waltham, MA, USA). PCR condition was 94℃ for 5 minutes for 1 cycle, and 94℃ for 45s, 60℃ for 45s, 72℃ for 120s for 30 cycles and 72℃ for 5 minutes for 1 cycle using Taq DNA polymerase (SolGent Co. Daejeon, South Korea). All of the coding exons (exon 2-exon 7) were analyzed by direct sequencing of PCR products using an ABI PRISM BigDye kit from Applied Biosystems. Forward and reverse sequences of PCR products were produced with an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
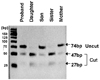
The result revealed that the proband had a heterozygous mutation of E297K (GAG→AAG, Glu→Lys, nucleotide 889 G A, exon 4) of CaSR gene (Fig. 1). The 74bp PCR product including Mnl I site was amplified using the primers CaSR_RFLP_F (CTCATCAAGGAGATTGTCCGG) and CaSR_RFLP_R (GAGGAGCTGGCCCAGGC). PCR amplification was carried out employing the same method mentioned above. The product of PCR amplification was digested with 1ul (5U) of Mnl I enzyme. The digestion occurred for 2 hours at 37℃, which was then electrophoretically resolved in components 2.5% agarose gel in 0.5X TBE. The result revealed that the proband had a heterozygous loss of an Mnl I site (Fig. 2).
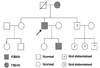
We performed biochemical and clinical evaluation of his family members. The plasma calcium of his 8-year-old son revealed 10.7 mg/dL, the inorganic phosphorus was 4.2 mg/dL and PTH was 56 pg/mL and random urine calcium 1.9 mg/dl, urine creatinine 82.0 mg/dL, the ratio of urine calcium creatinine clearance was 0.0013. The same mutation of codon 297 was found in his son and his 66-year-old mother. Biochemical and genetic analysis of his daughter and sister were normal (Fig. 3).
DISCUSSION
FBHH is a hypercalcemic syndrome usually characterized by as an asymptomatic and uncomplicated hypercalcemia and apparent normal longevity. The mode of inheritance is autosomal dominant with high penetrance, affecting both genders equally. The hypercalcemia is usually diagnosed at a young age, and it could be diagnosed as early as birth if screened for.4
FBHH is a rare disorder, but it is clinically important because it can be confused with primary hyperparathyroidism. The biochemical features of the two conditions are similar, but the former is benign while the latter can have serious clinical consequences with patients occasionally proceeding to parathyroidectomy.
The serum calcium level (total and ionized) is usually mildly elevated; values are indistinguishable from those of patients with primary hyperparathyroidism. Similar to hyperparathyroidism, serum phosphate levels are normal to slightly decreased, but serum magnesium levels are normal to slightly elevated.4-7 Abnormal calcium-sensing in FBHH is reflected in the inappropriately normal parathyroid hormone (PTH) levels in the overwhelming majority of patients with FBHH, whereas over 85% of patients with hyperparathyroidism have elevated PTH levels.8,9 In FBHH, 1,25-dihydroxyvitamin D levels have been reported to be normal.5 Urinary indices usually reveal relative hypocalciuria for the degree of hypercalcemia, pointing to excessively avid tubular renal calcium reabsorption.4,5,10 A calcium creatinine clearance ratio ([UCa × SCr]/[SCa × UCr]) below 0.01 has been suggested as cut-off values in patients with FBHH in the absence of other factors lowering urinary calcium excretion, whereas a ratio over 0.02 as with hyperparathyroidism.4,5,10 Kidney function, is normal, as well as urinary concentrating ability, in contrast to hyperparathyroidism.4,11
The calcium-sensing receptor (CaSR) is a cell surface protein that consists of 1078 amino acids and belongs to the superfamily of the seven-transmembrane domain, G protein coupled receptors.2 Over 90% of FBHH kindred's have their genetic abnormality linked to chromosome 3q21 - 24, the locus for the CaSR gene.3 Most mutations in the CaSR associated with FBHH are single point mutations (inactivating mutation) localized in either the region encoding the extracellular calcium-sensing domain, particularly exon 3 and 4, or the region encoding the signal-transducing domain, exon.7
Renal cell carcinoma often shows hypercalcemia, so we evaluated the relationship between renal cell carcinoma and hypercalcemia in this case. The radiologic findings revealed no evidence of distant metastasis including bone or local recurrence. The laboratory findings of the patient were not relevant to those in the case of malignancy due to elevation of PTHrP, such as hypercalcemia, hypophosphatemia, hypercalciuria, and decreased levels of parathyroid hormone. Von Hippel Lindau disease (VHL) is an autosomal dominant familial cancer syndrome.12 Affected patients are predisposed to develop central nervous system hemangioblastomas, renal cell carcinoma, and pheochromocytoma.12,13 To determine if a relationship existed between FBHH in this case and VHL, we analyzed the pedigree and chromosomes of the family and searched for any existing organ involvement. The findings demonstrated that there was no relationship between FBHH and renal cell carcinoma or between FBHH and VHL in this case.
Currently, more than 40 inactivating mutations in the CaSR gene have been identified in patients with FBHH.14-18 We report here a Korean family with FBHH carrying a mutation in the CaSR gene. The mutation was located in extracellular domain of CaSR as E297K mutation, which was already reported by Pollak et al. in 1993.2 The prevalence of E297K mutation is not reported, and this is actually the second case in English literature. The prevalence of CaSR gene mutation associated with FBHH is even largely unknown, but is estimated at 1 of 78,000, at least in Scotland.19
The mechanism of CaSR insensitivity by E297K heterozygous mutation was studied in human embryonic kidney cells. The E297K mutation leads to formation of heterodimers of CaSR compared to wild type homodimerization. As a result, extracellular calcium-elicited increase in cytosolic calcium is impaired about 63%.20 To our knowledge, it is the first case of FBHH confirmed with CaSR gene mutation in Korea, and the second case of E297K mutation in English literature.

 XML Download
XML Download