

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Nephrogenic diabetes insipidus (NDI) is a disorder that is inherited or acquired, and it is characterized by the loss of the ability to concentrate urine, and the insensitivity to arginine vasopressin (AVP).1 Most cases of congenital NDI (> 90%) are caused by a mutation in the AVP receptor 2 gene (V2R), and this is inherited as a X-linked recessive trait. The remaining cases are caused by mutation in the aquaporin 2 (AQP2) water channel gene with an autosomal recessive or dominant inheritance pattern.1-3 Patients with congenital NDI present in infancy or childhood with the various clinical symptoms of polyuria and polydipsia, and there are rare long-term sequelae such as growth retardation and urologic complications.4 Urologic problems in NDI have been reported as ranging from mild dilatation of ureter to severe hydronephrosis with neurogenic bladder.5,6 The diagnosis of congenital NDI with hydronephrosis is often confused due to this disease's similarity with acquired NDI of postobstuctive uropathy7-10 and the relatively rare incidence of congenital NDI that presents with hydronephrosis. Molecular analysis would be a useful diagnostic method, particularly for the unusual clinical presentation of congenital NDI. In this study, we describe two congenital NDI patients with mutations in the V2R gene and severe hydronephrosis.
CASE REPORT
Case 1
A 20-year-old man was admitted to Yonsei Medical Center, Seoul, Korea, because of the polyuria he had suffered from for several years. He has experienced polyuric symptom since childhood, however, he did not receive any specific evaluation except for intermittent medication for enuresis. On physical examination, he had normal blood pressure and no growth failure. His urine volume was more than 5,000 mL per day.
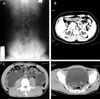
His blood laboratory results were: serum Na+ 145 mEq/L, Cl- 106 mEq/L, K+ 3.8 mEq/L, tCO2 27 mEq/L, BUN 7.9 mg/dL, and creatinine 1.1 mg/dL. The urinalysis was normal except for the low specific gravity (< 1.003). Serum and urine osmolality were 302 and 96 mOsm/kg H2O, respectively. An intravenous pyelogram and CT revealed a markedly distended bladder and bilateral hydronephrosis without any obstructive lesion of the ureter or urethra (Fig. 1A and 1B).
Case 2
A 21-year-old military soldier was admitted to the Armed Forces Capital Hospital because of maladaptation to the military services. He has had hard time coping in the military services due to his polyuria and nocturia that occurred everyday. He has urinated every 1 hour for several years, but, no specific evaluation or treatment had ever been done. On physical examination, he had normal blood pressure and a normal growth development. His urine volume was more than 10,000 mL per day. His blood laboratory results were: serum Na+ 154 mEq/L, Cl- 108 mEq/L, K+ 2.9 mEq/L, tCO2 30 mEq/L, BUN 17.1 mg/dL, and creatinine 1.6 mg/dL. The urinalysis was normal except for the low specific gravity (< 1.003). The serum and urine osmolality were 303 and 87 mOsm/kg H2O, respectively. Abdomino-pelvic CT showed a markedly distended bladder, bilateral hydroureter, and hydronephrosis without any organic obstruction (Fig. 1C).
Fluid deprivation test with AVP responsiveness
The patients stopped all medications for at least a week before the studies. Fluid deprivation test was performed with the study protocol being as described previously.11
Genomic DNA isolation from blood and PCR
DNA extraction from the whole blood was performed by using a Qiagen DNA mini kit (Qiagen Inc., Valencua, CA, U.S.A.). Four pairs of oligonucleotide primers were designed to allow the amplification of all three exons in the V2R gene (Table 1). PCR was performed using genomic DNA 100 ng, 1U of Taq polymerase, 0.4 uM each of the sense and antisense primers, 1.5 mM MgCl2 and 20 µmol/L dNTP in a volume of 50 L containing 1 × PCR buffer. The PCR conditions used were as follows (Table 1): Initial heating at 95℃ for 9 minutes, and this was followed by 35 cycles of denaturation at 95℃ for 45 seconds, annealing at the corresponding temperature for 45 seconds, and extension at 72℃ for 1 minute. And a final extension step at 72℃ for 7 minutes was performed. The PCR products were confirmed by ethidium bromide staining after electrophoresis in 2% agarose gel, and desired spots were cut out of the gel with a razor blade for DNA sequence analysis.
DNA sequence analysis
The PCR products were purified using a DNA purification kit (PCRquick-spin, Intron, Sungnam, Kyungki-do, Korea). Sequencing was preformed by the cycle sequence method with a DNA Sequencing Kit (Applied Biosystems, Foster city, CA, U.S.A.) in a thermal cycler (PCR 9600, Applied Biosystems, U.S.A.).
Fluid deprivation test
The ability to concentrate urine was impaired for both case 1 and 2 patients despite of the increase in serum osmolality. In addition, the urine osmolality remained below 100 mOsm/kg H2O even after desmopressin administration (Table 2).
Identification of mutation in the V2R gene
Mutation analysis was performed on the V2R gene of these patients. A missense mutation in each patients was identified by direct sequencing analysis (Fig. 2). In patient 1, a C to T transversion resulted in an early termination at amino acid 225 (Q225X). For patient 2, one base substitution of C for T was identified at the 809 nucleic acid in exon 2, which had Ser for Phe substituted at codon 126 (S126F).
DISCUSSION
While water permeability of the principal cells in distal nephron is relatively low in the basal state, AVP is secreted in response to various osmolar or volume stimuli and it binds to the V2R on the principal cells which leads to the movement of AQP-2 water channels from the intracellular vesicles into the apical plasma membrane, resulting in the increase in the water permeability.12,13 The mutations in the V2 receptor gene or the AQP-2 water channel gene can induce resistance to AVP which is a pathophysiologic characteristic of NDI. Congenital NDI is mostly inherited as an X-linked recessive trait that is caused by V2R mutation, and in minor cases, it is transmitted as an autosomal recessive form caused by mutation in AQP2 water channel gene. Although this disorder is caused by various mutations and various inheritances, the phenotypical features of the patients are identical.
In most cases, clinical symptoms with polyuria usually begin within the first week of life, however, these features are often not recognized in early infancy. In childhood or early adolescence, the affected patients have recurrent episodes of severe hypernatremia due to dehydration, and they also have non-specific symptoms such as anorexia, nausea, and fever. Unless this condition is treated appropriately, the recurrent episodes of dehydration can lead to growth disturbance and mental retardation in severe cases.14,15 In addition, there are urologic complications ranging from mild ureter dilatation to severe hydronephrosis, megaureter, and megabladder that develop due to the large urine volume in patients with congenital NDI.16-19
Differentiation of the congenital disease from acquired NDI, which is caused by long-standing neurogenic bladder or postobstuctive uropathy, may be difficult, because both diseases are occasionally accompanied with polyuria and hydronephrosis.7,9,10 Therefore, a tentative diagnostic methodology other than the clinical findings is mandatory to confirm congenital NDI. Ever since the V2R gene was cloned in 1992, gene mutation analysis has become the best confirmative method for congenital NDI.20,21 As a result, approximately one hundred mutations in the V2R gene have been reported in the literatures, however, there has been no report on the V2R mutation for any cases of Korean congenital NDI with hydronephrosis. Hence, we report here on two Korean patients with congenital NDI that presented with bilateral hydronephrosis; one patient had the Q225X missense mutation and the other patient had the S126F mutation.
It is obvious that the Q225X missense mutation is responsible for causing the AVP unresponsiveness in the distal collecting tubule because the mutation results in the premature termination of the amino acid in V2R gene, and the mutated products are short compared to the intact V2R.22 In addition, the S126F mutation has previously been reported in Canadian patients,22 and this may be one of the hot spots for nucleotide substitutions. This amino-acid substitution probably induces conformational changes of the V2R gene, resulting in the loss of function of the V2R protein.
In conclusion, we report here on two cases of congenital NDI with bilateral non-obstructive hydronephrosis that were diagnosed by fluid deprivation tests and the diagnosis was confirmed by V2R mutation analysis. Since the structural changes of the urinary tract and growth retardation are reversible when the treatment is initiated as soon as possible, genetic counseling and evaluation for the high-risk family members is highly recommended.



 XML Download
XML Download