INTRODUCTION
Cervical cancer and precancerous cervical lesions are a major problem in women's health. Clinical, molecular and epidemiological investigations have identified HPV as the major cause of cervical cancer and cervical dysplasia. Virtually all cervical cancers contain high-risk HPV genes, most commonly types 16, 18, 31, and 45.1 HPVs infect epithelial cells of skin and mucosal surfaces.2 To date, at least 80 distinct types of HPV have been isolated from various human tissue biopsies. A subset of these HPVs is sexually transmitted and causes a spectrum of diseases in the male and female genital tract.3 These HPVs can be categorized according to their potential to cause malignancy as low risk, intermediate risk or high risk. HPV types 6 (HPV-6) and 11 (HPV-11), for example, are associated with low-risk disease, such as genital warts. On the contrary, high-risk HPVs, such as HPV-16, -18, -31, -33 and -45, are associated with high-grade intraepithelial lesions and invasive cancer. Among the high-risk strains, HPV-16 and -18 are the most closely associated with cervical carcinoma. HPV-16 DNA has been found in more than 50% of squamous cell carcinomas, while HPV-18 DNA has been found in more than 50% of adenocarcinomas.2
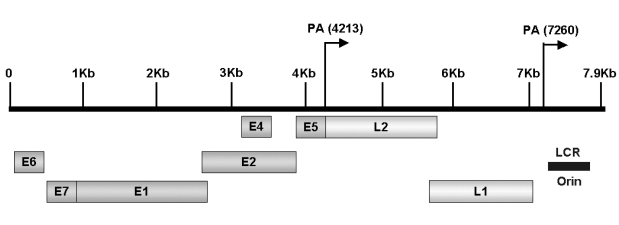
HPVs comprise a large family of double-stranded DNA (ds DNA) tumor viruses whose genome is organized into three regions: early genes (E1 to E7), late genes (L1 and L2) and the non-coding long control region (LCR) containing a variety of cis elements4 which regulate viral replication and gene expression (Fig. 1). The LCR possesses numerous binding sites for many repressors and activators of transcription. Both E1 and E2 have been shown to be essential for the replication of the origin of HPV. The E1 and E2 encode proteins which are essential for extrachromosomal DNA replication and completion of the viral life cycle. The E2 is a DNA binding transcription factor interacting with ACCN6GGT motifs in the viral LCR.5 The E2 encodes two proteins which inhibit and activate the early region, respectively. A hallmark of HPV-associated cervical carcinoma is the loss of this E2 protein expression.6 In addition to modulating viral gene expression, the E2 proteins are associated with the viral helicase E1. This interaction is necessary for efficient recognition of the origin and viral genome replication.7,8 An E4 protein is expressed in the later stages of infection after completion of the virion assembly. The E4 is considered to play an important role in the maturation and replication of the virus.9 The E4 protein also induces the collapse of the cytoplasmic cytokeratin network in human keratinocytes. This situation may facilitate the release of virions from infected cells.10 Expression of the viral E1-E4 proteins are lost during a malignant progression, but not in premalignant lesions. Expression of HPV-16 E1-E4 in cell cultures is high and causes cell cycle arrest at the G2 phase by sequestration of cyclin-dependent kinase 1 (Cdk1)/cyclin B1 in the cytokeratin network. This prevents the accumulation of active Cdk1/cyclin B1 complexes in the nucleus resulting in inhibition of mitosis. In another report, HPV-16 E1-E4 mutants (T22A, T23A) which do not bind cyclin B1 or alter its intracellular location failed to induce G2 arrest.11 Meanwhile, an E5 open reading frame (ORF), is often deleted in cervical carcinoma cells. This indicates that it may not be essential in maintaining the malignant transformation of the infected cells.12 The E5 interacts with various transmembrane proteins such as receptors of the epidermal growth factor (EGF),13 platelet-derived growth factor β (PDGFβ)14 and colony stimulating factor (CSF). The E5 protein has transforming activity in HPV-16 infected cells. In the protein-encoding regions, E6 and E7 ORFs are considered to play major roles in HPV pathogenesis. These two genes encode for oncoproteins allowing replication of the virus, immortalization and transformation of the host cells.15-18 Late region units, L1 and L2, encode for viral capsid proteins during the late stages of virion assembly (Table 1). The protein encoded by L1 is highly conserved among different papillomavirus species. Therefore, antibodies against the Bovine Papillomavirus (BPV) have been used to identify HPV capsid proteins in human tissues. The minor capsid protein encoded by L2 has more sequence variations than that of the L1 protein; hence, antibodies against the L2 protein have been a source of antigens for specific types of HPV antibodies.19
THE ROLE OF THE HUMAN PAPILLOMAVIRUS E5 IN CERVICAL CARCINOGENESIS
HPV-16 E5 protein
The HPV-16 E5 protein is a small (84 aa) hydrophobic membrane protein located just downstream of the E2 open reading frame.20 The gene is not well conserved at the DNA level among HPVs or animal viruses, although there is conservation of some properties, as the proteins are all highly hydrophobic and membrane-bound.20 The E5 proteins from both animal and human sources can transform mammalian cells with varying degrees of efficiency. The protein is membrane-bound and distributed predominantly in the endoplasmic reticulum, the Golgi and the cytoplasmic membrane.21 The E5 protein is hydrophobic and presents as a dimer even in reducing conditions. The E5 protein is difficult to detect because the protein is insoluble and generation of good antibodies against E5 are limited. Antigenic peptide antibodies against the N-and C-terminal have been produced successfully. Fragment deletion of the E5 ORF in cervical carcinoma cells indicates that E5 does not play an essential role in maintaining the malignant phenotype. This allows the presence of the E5 protein to be used as a prognostic predictor marker.22 The HPV-16 E5 protein contributes to cellular transformation by increasing a mitogenic stimulus from growth factor receptors to the nucleus. In this transformation mechanism, the E5 protein co-localizes with an anti-apoptotic Bcl-2 protein.23 The E5 protein of BPV impairs the synthesis and stability of the major histocompatibility complex (MHC) class I and prevents their transport to the cell surface due to retention in the Golgi apparatus. The HPV-16 E5 protein also causes the retention of MHC (HLA) class I in the Golgi apparatus and impedes their transport to the cell surface, which is rescued by treatment with interferon. Unlike BPV E5, HPV-16 E5 does not affect the synthesis of HLA class I heavy chains or the expression of the transporter associated with antigen presentation (TAP). These results show that down-regulation of the surface MHC class I molecules is common to both BPV and HPV E5 proteins.
Transformation and immortalization properties
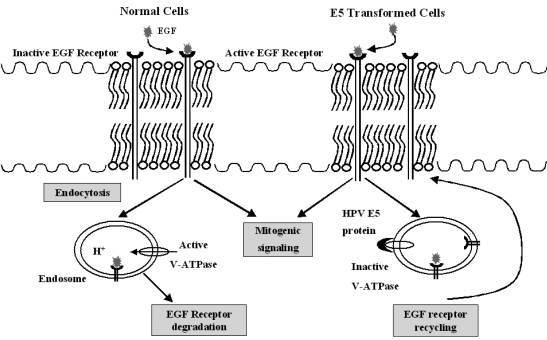
There are well recognized cellular targets for the E5 protein: PDGFβ-R and the 16-kDa subunit of the vacuolar H+-ATPase.24 The original E5-receptor interaction was described for BPV type 1 E5. The E5 viral protein was found to stabilize an epidermal growth factor receptor (EGFR) in the presence of the ligand. The ligand-receptor complex was not broken down and a hyper-stimulating signal was observed.25 The BPV-1 E5 was found in a complex with the PDGFβ receptor (PDGFβ-R)26 and could activate PDGFβ-R in the absence of the ligand PDGFβ. The E5-PDGFβ-R interaction occurs during the natural history of bovine urinary bladder tumors, suggesting an important role for E5 in carcinogenesis.14 However, the PDGFβ-R is not found on human keratinocytes. The EGFR is the major receptor in keratinocytes. The HPV E5 proteins appear to affect the activity of growth factor receptors and their signaling pathways. The interaction of E5 proteins with a subunit of vacuolar adenosine triphosphatase (ATPase) may contribute to a transformation.12 In order to elucidate this E5 oncogenic activity, the significance of the interaction between E5 and EGFR has been studied. Enhanced activation of the signaling cascade by E5 is EGFRdependent. In the presence of the E5 protein, the EGFR number is increased.13 This effect is due to an impaired down-regulation of receptors and recycling of those to the cell membrane in E5-expressing cells. An ubiquitination of the activated EGFR plays a role in this down-regulation (Fig. 2). A c-Cbl is a ubiquitin ligase which associates with the activated EGFR and targets it for degradation. HPV-16 E5 may, at least in part, upregulate EGFR-mediated signal transduction by inhibiting the interaction of the c-Cbl with the EGFR, then decreasing the c-Cbl-mediated degradation of the EGFR.27 Moreover, this enhanced receptor recycling is mediated by E5 binding to a 16-kD subunit of the ATPase proton pump located in the endosomal membrane. This binding can be demonstrated in vitro.27 In vivo data also revealed the E5 protein predominantly existing in the cytoplasmic membrane region of E5-transfected cells and the HPV-16 infected tissue, with the E5 protein binding to the 16-kD molecule. In contrast, one study reported that EGF treatment increased EGFR phosphorylation instead of the receptor number in E5-expressing cells.26 This result suggested that E5 could form a complex with EGFR to induce the phosphorylation of EGFR. However, other researchers could not demonstrate an E5-EGFR complex-induced signaling pathway. An EGFR-independent pathway has also been proposed to function in the E5 protein-activated signaling cascade. The HPV-16 E5 modulates the sorbital-dependent activation of mitogen-activated protein (MAP) kinase p38 and ERK1/2 in human keratinocytes through an EGF-independent mechanism.28 Taken together, E5-activating signaling cascades appear to be mediated via either a receptor-dependent or receptor-independent pathway.
The HPV-16 E5 can transform immortalized rodent fibroblasts and the frequency of transformation is increased in the presence of EGF but not PDGF. The HPV-16 E5 protein is unable to form transformed foci on cell monolayers and therefore is considered to have only moderate transforming activity, unlike the BPV-1 E5 gene, which can form transformed foci on rodent monolayer cultures. It should be remembered that the BPV-1 E5 protein is the major transforming protein, unlike HPVs' E5. The HPV-16 E5 protein is unable to immortalize primary human keratinocytes,29 however, the E5 induces cell proliferation and extends the life span of cells predisposed with activated EFGR. In addition, the full length HPV-16 genome with a premature stop codon in the E5 gene is only able to immortalize keratinocytes at 10% of the frequency of the wild type genome, suggesting that E5 plays a complementary role in the immortalization process.30
E5 and signal transduction
The HPV-16 E5 protein can activate EGFR, which in turn can initiate diverse biochemical events that ultimately render the over-expression of a variety of proto-oncogenes.31 The HPV-16 E5 protein might activate MAP kinases via two different pathways, one for the protein kinase C (PKC-dependent), the other for the receptor tyrosine-kinase mediated (PKC-independent) pathway.28 The E5 could stimulate the nuclear oncogenes, such as c-jun, junB and c-fos.32 The c-jun nuclear oncogene is activated by HPV-16 E5 via PKC and ras-dependent pathways that transmit signals to the nucleus. The E5 activation of c-jun is through an activator protein-1 (AP-1) binding site and the E5-activation of c-fos is via a nuclear factor-1 (NF-1) binding site in the nucleus. Since several binding sites of AP-1 and NF-1 are located in the regulatory region of the HPV-16 DNA, these transcription factors can activate the enhancer of HPV-16, and the E5 may potentiate viral gene expression via the activation of AP-1 and NF-1.20 Thus, the E5 protein transactivates viral genes and thereby increases the viral E6/E7 gene expression and regulates E7-mediated cell transformation. In conclusion, the E5 protein may play a role in cell transformation by regulating the expression of other cellular and viral genes. The E5 protein could down-regulate the expression of the p21(WafI/SdiI/CipI) tumor suppressor gene in NIH 3T3 cells and in immortalized human keratinocytes,33 which could promote cell proliferation. This observation also suggests a role for E5 in augmenting or supplementing the function of E6 and E7 during immortalization of human genital keratinocytes in the course of primary infection. Colony numbers were higher in primary keratinocytes infected with the full-length HPV-16 genome than in those with mutated E5 gene-containing HPV-16 genomes. This finding demonstrates that the E5 protein can act in cis to increase the efficiency of cellular immortalization by the E6/E7 protein. In addition, mutations in E7 of the HPV-16 genome, which inhibits the binding of pRb, could not abrogate HPV-16-induced immortalization of primary human keratinocytes. Moreover, intradermal injection of cottontail rabbit papillomavirus DNA, which has mutations in the E7 gene sequences critical for the binding of pRb, could still induce papilloma formation in rabbits. Hence, the repression of the p21 gene by the E5 protein may complement the altered pRb binding activity of the mutated E7. Moreover, suppression of the p21 gene by E5 may facilitate the activation of Cdk4-cyclin D complexes, which are known to phosphorylate pRb and inactivate the Rb-checkpoint control. It remains uncertain whether the E5 can supplement the E6 transformation activity. The E6 protein targets the tumor suppressor protein p53, resulting in p53 degradation and contributing to malignant transformation. Although p53 can activate the gene expression of p21, it is unclear whether repression of the p21 gene expression by HPV is in a p53-dependent manner.34 Therefore, investigations are still necessary to decipher the complicated networks existing among E5, E6, p53, and p21.
ONCOGENESIS INDUCED BY HPV-16
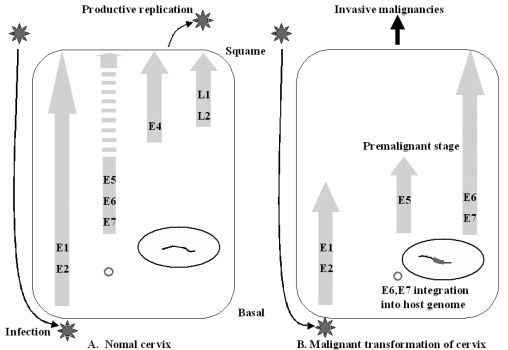
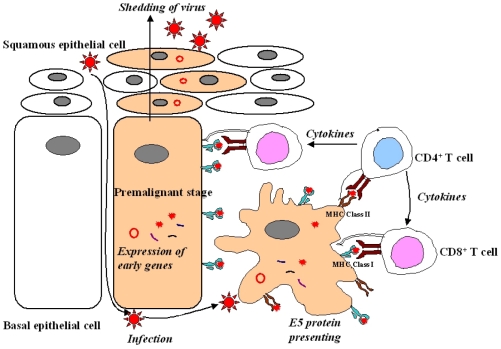
In cervical neoplasia, high-risk HPV infection occurs predominantly in either basal or parabasal cells of the transformation zone epithelium.35 The HPV virion infects basal cells of the stratified mucosal epithelium at the junction between the vagina ectocervix (Fig. 3). Limited viral replication is accompanied by expression of the early proteins E1 and E2 in the basal layer. In more distal layers, E6 and E7 proteins are expressed. These proteins promote cell proliferation and delay differentiation. As infected cells differentiate into squamous cells, the E4 protein and the late proteins L1 and L2 are expressed. The viral capsid is shed into the genital tract within desquamated epithelial cells. The E5 protein is likely to play a role during the premalignant stage.3 Rarely is the circular HPVs genome linearized randomly and integrated into a host genome. During the HPV DNA integration, the viral genome usually breaks in the E1/E2 region, resulting in the loss of E1 and E2 regions. The E2 encodes proteins which inhibit the transcription of the E6 and E7 regions. The loss of E2 leads to uncontrolled and increased expression of the E6 and E7 oncogenic proteins. The increased expression of E6 and E7 meanwhile has led to the malignant transformation of host cells and to tumor formation.36 The viral particles, composed of major (L1) and minor (L2) capsids, access the target cells and facilitate the entry of the viral DNA, probably following minor local trauma. Subsequent viral protein expression correlates with differentiation in the spinous layers. Initially, the immediate early proteins E1, E2 and E5 can be detected. Each E1 and E2 ORF encodes DNA-binding proteins which are required for maintaining a stable viral episome. The E2 is involved in the positive and negative regulation of viral gene expression through a specific interaction with the early promoter found in the upstream regulatory region. In the lower spinous layers, the E6 and E7 proteins are also expressed with the other early proteins. They regulate cell proliferation and interfere with the host cell cycle control mechanisms to activate cellular DNA synthesis. This appears to be essential for viral vegetative DNA replication. In the upper spinous layers, vegetative DNA amplification and virus assembly occurs with the L1 and L2 protein expression. Although the E4 is encoded within the early region, it is also largely restricted to the upper spinous layers.37 The precise role of E4 is unknown, however, it is known to interact with the keratin intermediate filaments in cultured epithelial cells. Mature virions are released from exfoliating cells. In HPV types associated with a malignant transformation, it is clear that E6 and E7 are the predominant transforming proteins, although E5 can also show oncogenic potential. The major mechanism by which oncogenic HPV E6 and E7 contribute to the development of cervical cancers is due to a functional interaction with p53 and Rb, respectively. These proteins play a pivotal role in the negative regulation of the growth. The E6 from oncogenic HPVs binds to p53 with high affinity, resulting in a loss of p53-dependent functions including G1 arrest and apoptosis.38 The consequence of E7 binding to Rb is to prevent Rb binding to and sequestration of E2F transcription factors, leading to a disruption of the cell cycle control.39 In the initial infection, HPV is present as an episome. However, in the majority of more advanced lesions and invasive tumors, HPV is integrated into the host genome. The E6/E7 transcripts from integrated HPV genomes have increased stability as compared with episomally derived viral mRNA, and there is an increased expression in more severe lesions and cancers. This may result from a disruption of the E2 gene during integration. Following integration, viral particles can no longer be produced, but continued E6 and E7 activity prolongs the cell cycle, leading to the loss of effective DNA repair mechanisms. This provides opportunity for the accumulation of genetic changes in a multi-step process that result in the development of cancer. Therefore, numerical and structural chromosomal changes in cervical cancer are common, and allelic losses have been observed in many different chromosomal regions.40 The E5 is expressed in earlier stages of neoplastic transformation on cervical epithelia during viral infection. Since early lesions usually contain fewer tumor cells, a host immune response may have a higher chance to eradicate tumor cells by tumor-specific cytotoxic T lymphocytes (CTL) in premalignant lesions than in invasive cervical cancers (Fig. 4).
OTHER ONCOGENIC VIRAL PROTEINS, E6 AND E7
HPV-16 E6 protein
The HPV-16 E6 protein is a 151-amino acid protein, containing two zinc finger domains. The E6 induces several important changes in a host cell that impact both the normal viral life cycle and the process of immortalization. The E6 alone is not capable of immortalizing primary human foreskin keratinocytes (HFK), but can efficiently immortalize human mammary epithelial cells (HMEC).9 However, the E6 acts with the E7 to immortalize HFK. Expression of the E6 as a transgene in mice leads to hyper-proliferation of epithelial layers, loss of differentiation in epithelial layers, and formation of benign and malignant tumors. The E6 is one of the earliest expressed genes during the HPV infection. It alters the cellular environment and confers it more amenable to production of new viral particles. This includes blocking apoptosis through p53 degradation,41 altering transcription of cellular genes through interaction with p300, and increasing cellular lifespan through increased telomerase activity.42 These functions serve to facilitate the viral life cycle.
HPV-16 E7 protein
The HPV E7 ORF has the major transforming activity. The HPV-16 E7 is a nuclear protein of 98 amino acids with casein kinase II phosphorylation sites at serine residues 31 and 32.43 The protein is divided into domains based upon homology to the adenovirus E1A. The domains are: CR1 consisting of amino acids 1-20, CR2 containing residues 21-39 and CR3 composed of amino acids 40-98. The E7 dimerizes via a zinc-finger motif in the CR3 region essential for proper protein folding. Numerous interactions between E7 and cellular proteins have been described.9 Many of these interactions involve factors regulating cell growth, especially the transition from the G1 to S-phase in the mitotic cycle. The E7 protein has been demonstrated to be associated with the retinoblastoma tumor suppressor protein family (Rb, p107, p130), histone deacetylases (HDAC), AP-1 transcription factors, cyclins, Cdks and cdk inhibitors.44 These associations likely contribute to the ability of the E7 to induce cellular proliferation, immortalization and transformation.
CURRENT HPV VACCINES
The well-defined virologic, genetic and pathological progression information on HPV, from initial infection to lesion formation to malignant tumor formation, and the limited number of well-defined viral antigens, provide unique opportunities to evaluate interventions with antigen-specific immunotherapy at various stages of HPV infection and tumorigenesis. Conceptually, two different types of HPV vaccines can be designed, one for prophylactic (preventive) vaccines preventing HPV infection and the other for therapeutic (curative) vaccines inducing regression of established HPV infection and its sequelae.
Prophylactic HPV vaccines
Prophylactic HPV vaccine development has been complicated by the lack of animal models for the genital mucosatropic HPV types and by difficulty in propagating the virus in culture. These difficulties have been partially overcome by using cutaneous and mucosal administration of animal papillomaviruses and by the development of virus-like particles (VLPs) and pseudovirions.45 Yet, these models do not adequately simulate sexual transmission. When the papillomavirus major capsid protein L1 is over-expressed in mammalian, insect, yeast or bacterial cells, capsomeres are spontaneously assembled to form VLPs which are devoid of the oncogenic viral genomes. Parenteral injection of these VLPs elicits high titers of serum-neutralizing antibodies and protective immunity, as shown from several animal papillomavirus model systems with experimental challenge of infectious virus. Although VLP vaccination provides a host immunity from the experimental inoculation, it is unclear whether this extends to protection against natural transmission of genital HPV.46 To completely prevent sexual transmission of genital HPV infection, neutralizing antibodies must act at mucosal surfaces, which are the natural site of infection.47 Antibodies are produced by the plasma B cell passing into genital secretions and by local plasma cells. The plasma cell precursors which migrate to the genital tract are derived primarily from mucosal lymphoid tissues and predominantly secrete IgA. Induction of these cells requires direct immunization of the mucosa-associated lymphoid tissue. In several experimental systems, nasal instillation was found to be the most effective method of immunization to generate specific antibodies in genital secretions in mice and monkeys. More recently, oral vaccination with the HPV VLPs in mice has been shown to induce systemic virus-neutralizing antibodies, suggesting that the HPV VLPs may be antigenically stable in the environment of the gastrointestinal tract. These studies provide the possibility of vaccinating large populations with the HPV VLP without using syringes. Local, sustained production of secretory IgA and/or specific IgG is likely required for long-lasting sterilizing immunity. In previous studies, systemic immunization of mice with the purified HPV VLPs induced no detectable mucosal IgA antibodies and low titers of IgG. Furthermore, low titers of VLP-specific IgG, and no IgA, were detected in cervicovaginal lavage of parenterally immunized monkeys.48 Nasal immunization using HPV-16 VLPs has been shown to induce significant and sustained titers of HPV-16 neutralizing antibodies in both serum and mucosal secretions of mice. The promising results generated in preclinical animal studies have led to phase I/II clinical trials using the HPV-16 VLP vaccine delivered intramuscularly. Recently, Liu et al.49 showed that the HPV-16 L1 protein could be expressed in transgenic tobacco plants and that the expressed protein possessed the natural features of the HPV-16 L1 protein, implying that the HPV-16 L1 transgenic plants could potentially be used as an edible vaccine. These studies suggest the possibility of vaccinating large populations with the HPV VLPs without using syringes. In recent clinical data, intramuscular vaccination of women with the HPV-16 VLPs was found to induce significant antibody titers in cervical secretions.50 A comparison of nasal spray versus aerosol for mucosal delivery of 50 µg of HPV-16 VLPs to humans revealed that aerosol was more effective for inducing an antibody response, with IgG found predominantly in serum and both IgG and IgA found in cervical secretions. A recent clinical trial of HPV-16 L1 VLPs shows for the first time that a vaccine strategy could be implemented in humans to prevent HPV-16 infection and HPV-16-associated premalignant lesions.51 Taken together, using VLPs for prophylactic vaccination prevents HPV infection and disease.
Therapeutic HPV vaccines
The development of a second-generation vaccine that will provide a T-cell response to eliminate infected cells is promising. Therapeutic vaccines should induce specific cell-mediated immunity which prevents the development of lesions and eliminates pre-existing lesions or even malignant tumors. Theoretically, the induced antigen (Ag)-specific cellular immunity can directly target HPV viral gene products, the HPV-induced cellular products, or a combination of both. The L1 and L2 capsid proteins are unlikely to be suitable targets for therapeutic vaccine development because these proteins are not detectably expressed in basal epithelial cells of benign lesions or in abnormal proliferative cells of premalignant and malignant lesions.3 Although a previous study indicated that vaccination with VLPs may generate a capsid protein-specific CTL response, such a response against L1 or L1/L2 VLPs alone would not likely result in a significant therapeutic effect.52 Since E6 and E7 are consistently expressed in most cervical cancers and their precursor lesions, but are absent from normal tissues, these viral oncoproteins represent promising targets for the development of Ag-specific therapeutic vaccines for HPV-associated cervical malignancies and their precursor lesions. While most tumor-specific Ags are derived from normal or mutated proteins, E6 and E7 are completely foreign viral proteins. Therefore, these proteins harbor more antigenic peptides/epitopes than an altered cellular protein. Furthermore, since E6 and E7 are required for the induction and maintenance of the malignant phenotype of cancer cells, cervical cancer cells are unlikely to evade an immune response through Ag loss.53 Studies using modified adenovirus (AdV)-expressing HPV-16 E6 or E7 for vaccination or for ex vivo preparation of dendritic cell (DC)-based vaccines have demonstrated an enhancement of Ag-specific T-cell immune responses and anti-tumor effects.54 A replication-defective adeno-associated virus (AAV) encoding HPV-16 E7 fused to heat-shock protein 70 was found to induce CD4- and CD8-dependent CTL activity and anti-tumor effects in vitro.55 Peptide vaccines have the advantage of safety and ease of production, however, their weak immunogenic properties and the need for human leukocyte antigen (HLA) matching must be overcome. Because 40% of Caucasians carry the HLA-A2 alleles, HPV-16 E7 peptides presented by this allele have been the immunogen in several phase I/II clinical trials.56 Whereas peptide vaccines exhibit MHC restriction, protein-based vaccines can bypass this restriction and thus are less dependent on the patient's HLA type. The HPV-6 L2 fused to the E7 protein has been successfully tested for the clinical treatment of genital warts.57 The potency of HPV-16 E7 peptide-based vaccines may be further enhanced through the use of adjuvants, fusion proteins, or anchor-modified peptide epitopes. Chu et al.58 demonstrated that adjuvant-free immunization of C57B1/6 mice with heat shock protein E7 (hspE7) protected the animals against challenge with an E7-expressing murine tumor cell line (TC-1), and also protected against a rechallenge with higher doses of TC-1 cells. Similarly, immunization of tumor-bearing mice with hspE7 led to tumor regression, protection from re-challenge and long-term survival. Tumor regression after hspE7 immunization appeared to be dependent on CD8+ T-cell activity but independent of CD4+ T-cells. More recently, in a single-stage phase II study, DNA vaccines allowed for sustained antigen presentation as MHC-peptide complexes as compared with peptide or protein vaccines.59 Due to the weak intrinsic potency of naked DNA vaccines, various strategies have been developed to enhance their immunogenicity. To this end, recent research has used a method to prolong in vivo DC survival. Co-administration of the E7-containing DNA with anti-apoptotic proteins (Bcl-xl, Bcl-2) enables the DNA vaccine to enhance E7-specific immune responses, tumor treatment and DC survival.59 The DCs are the most potent antigen presenting cells (APCs) in the immune system and techniques have been developed for isolation, cultivation and Ag loading on MHC of DC. Santin et al.60 demonstrated that recombinant, full-length, E7-pulsed, autologous DCs could elicit a specific CD8+ CTL response against autologous tumor target cells in three patients with HPV-16 or HPV-18-positive cervical cancer. Their results demonstrated that full-length, E7-pulsed DCs could induce both E7-specific CD4+ T-cell proliferative responses and strong CD8+ CTL responses capable of killing autologous HPV-infected cancer cells in cervical cancer patients. Transduction of tumor cells with genes encoding co-stimulatory molecules or cytokines may enhance immunogenicity, leading to T-cell activation and anti-tumor effects after vaccination. Vaccines using HPV-transformed tumor cells transfected with cytokine genes, such as interleukin (IL)-12, IL-2, or granulocyte macrophagecolony stimulating factor (GM-CSF), can generate strong anti-tumor effects in mice.51
HPV vaccines targeting E5
The E5 is expressed in precancerous stages of cervical epithelium during HPV infection. Since precancerous lesions usually contain fewer cells than the invasive malignancies, it can be speculated that early immunological intervention might offer a chance to eradicate tumors more efficiently while still in a premalignant stage. Furthermore, cells in more advanced stages are found to have very low expression levels of MHC class I and II mRNA, which may consequently hamper the presentation of tumor Ag and lead to decreased immunosurveillance. Some reports demonstrated that lymphocyte proliferation in response to the HPV-16 E5 is inversely proportional to the severity of the squamous intraepithelial neoplasia lesions (SILs).61 Hence, in the E5-expressed precancerous lesions such as SILs and condyloma, using E5 as a vaccine target might be a reasonable strategy to prevent these from progression toward invasive cervical cancers. One investigation demonstrated that a single muscular injection of recombinant AdV encoding the HPV-16 E5 gene into mice with a TC-1 tumorigenic stable cell line engineered to express the E5 protein could reduce tumor growth in the lesions containing the E5 gene.62 The potency of the E5 vaccine might be exerted via CD8+ rather than CD4+ T-cells. This indicates that the E5 might be identified as a tumor Ag by the immune system. Recently, HPV-16 E5 25-33 peptide plus CpG oligodeoxynucleotides (ODN) was shown to be a superior vaccine as compared to the AdV-based E5.63
DISCUSSION
The HPV type 16 is the predominant type of virus identified in cervical cancers. It encodes three transforming oncogenes, E5, E6, and E7. The HPV genomes are commonly found to be integrated into the host genome. Therefore, the E6/E7 genes remain intact in the host genome, E5 is lost or, if present, under-expressed as compared with E6/E7. This suggests that E5 may play a critical role in the genesis of cervical cancer but less of a role in its persistence or progression. The E5, E6, and E7 are unique tumor Ags and may serve as ideal Ags to be targeted as a tumor vaccine. Most of the evidence for HPV Ag-directed immunotherapy against cervical cancer comes from the experimental and natural papillomavirus-associated tumors that can be controlled by immunization with E6/E7 Ags. In this review paper, we address the potential of the E5 protein as a tumor Ag inducing E5-specific CD8+ T-cells to eradicate tumor growth. In vivo studies demonstrate E5 expression soon after infection, with both mRNA and protein detectable in low-grade squamous intraepithelial lesions. The prevalence of E5 mRNA is increased with progressing severity of the disease. Therefore, when E5 is expressed in earlier stages of neoplastic transformation of the cervical epithelium due to viral infection, the early lesions usually contain fewer tumor cells. The immune effector cells in premalignanant lesions may eradicate tumor cells more efficiently than in the invasive cervical cancers. The CTL activated by the E5 protein will effectively kill tumor cells. Therefore, the E5 is a better target gene than other HPV-16 genes to eliminate tumor cells in the early onset stages of the disease.