

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The central nervous system (CNS) has been considered as an immune-privileged site. However, recent studies have shown that resident cells of the CNS, including astrocytes, microglia, oligodendrocytes, and neurons actively participate in ongoing immune responses in the CNS.1-3 Astrocytes are the most abundant glial cell type in the CNS and have been shown to produce and respond to cytokines. Such cytokines include tumor necrosis factor (TNF)-α, interleukin-1 (IL-1), granulocyte macrophage colony-stimulating factor (GM-CSF), transforming growth factor (TGF), IL-10, and IL-12 as well as chemokines.4-8 Some of these cytokines, such as TNF-α, have shown involvement in immune-mediated CNS disorders, including multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE). They are involved in ways that promote the infiltration of inflammatory cells into the CNS, intracerebral immune responses, cytokine production, astrogliosis, and demyelination.9-11 However, several experiments have demonstrated that TNF-α has neuroprotective actions. The dual action of TNF-α in the CNS may be determined by which types of signaling pathways, proapoptotic or antiapoptotic, are activated through TNF-α and TNF receptor interaction.
The induction of various cellular responses mediated by TNF-α is initiated by its interaction with two distinct cell surface receptors of approximately 55kDa (TNFR1) and 75kDa (TNFR2).12 While the extracellular domains of these two receptors exhibit some sequence homology, their intracellular or cytoplasmic domains appear quite dissimilar.13,14 This is thought to be indicative of the existence of distinct signaling pathways.
Recent studies have indicated that TNFR1 mediates the signals required for cytotoxicity and other TNF-α activities, including antiviral activity, fibroblast proliferation, and the induction of NF-κB, while TNFR2 is involved in signaling the proliferation of thymocytes, cytotoxic T cells, and human mononuclear cells.15-17
Several cytokines have been shown to regulate TNFR1 and TNFR2 expression in a variety of cells.18-21 The modulation of TNFR expression by inflammatory cytokines may represent a means of controlling the responsiveness of TNF-α.
TNF-α has multiple effects on astrocytes, such as increasing intracellular Ca2+ and depolarization, and inducing multiple cytokines and adhesion molecules.22 Upon its activation, TNF-α induces TNF-α secretion in an autocrine fashion.23 Moreover, TNF-α has been shown to induce cell proliferation in astrocytes.24,25
Conflicting results exist regarding the expressions of TNFR1, TNFR2, and their functions in astrocytes. Alanguez et al. reported that murine astrocytes express only TNFR1.26 However, a recent study found that TNF-α can induce TNFR2 gene expression, and provided a possible explanation of the proliferative effect of TNF-α in rat primary astrocytes.27
Little is known about the cell surface expressions of TNFR1 and TNFR2, and about their functions in human astrocytes. Moreover, no study has yet examined the regulations of TNFR1 and TNFR 2 expression by cytokines in human astrocytes.
In this study, we determined the expression levels of TNFR1 and TNFR2 in human fetal astrocytes. We also examined the effects of the inflammatory cytokines IFN-γ, IL-1β, TNF-α and bacterial LPS on the expressions of TNFR1 and TNFR2 mRNA in human fetal astrocytes. Soluble TNFR1 and TNFR2 were studied using an enzyme-linked immunosorbent assay (ELISA). Furthermore, we have tried to identify the receptor subtype involved in the induction of NF-κB activation and TNF-α production in human fetal astrocytes.
MATERIALS AND METHODS
Cell cultures
Human fetal astrocyte cultures were prepared as previously described.28,29 Briefly, human fetal brain tissue from therapeutically aborted fetuses of gestational ages between 16 and 24 weeks was minced and passed through a 19-gauge needle. Cells were plated in Dulbecco's modified Eagle medium (Gibco) containing 10% fetal bovine serum (Gibco), 2 mM glutamine, non-essential amino acids, penicillin/streptomycin and antimycotics. Cells were passaged for three to six weeks prior to use. Astrocyte purity was assessed by staining for glial fibrillary acidic protein (GFAP, Boehringer Mannheim), an astrocytespecific protein; the astrocyte cultures were composed of more than 95% GFAP (+) cells. Primary cultures from at least 10 different donors were used for these studies. Each experiment was repeated at least three times with cells from different donors. THP-1 human monocytic cell lines were obtained from ATCC and cultured in DMEM containing 10% fetal bovine serum (Gibco).
Reagents
Lipopolysaccharide and human recombinant IFN-γ were purchased from Sigma, and human recombinant IL-1β and TNF-α were obtained from Promega and Endogen, respectively. Neutralizing anti-human TNFR1 and anti-human TNFR2 antibodies were purchased from R&D Systems, as were ELISA Kits for soluble TNFR1 and TNFR2.
RNA isolation and competitive RT-PCR
Total cellular RNA was isolated from confluent monolayers of human fetal astrocytes that had been incubated with various cytokines. RNA was isolated using a Trizol reagent (Gibco), and the amount of RNA obtained was determined by spectrophotometry.
Reverse transcription was performed using 1 µg of total RNA with oligo (dT) 12-18, AMV reverse transcriptase (Promega), RNase inhibitor (Promega) and dNTP mix in a final volume of 20 µL. The mixture was incubated at 42℃ for 1 h and the reaction was terminated at 95℃ for 5 min. RT-PCR was performed with an automatic thermocycler (Hybaid). The following primers were used: 5'-AAATGGGTCAGGTGGAGATC-3' (forward), 5'-GGCTTAGTAGTAGTTCCTTC-3' (reverse) for TNFR1; 5'-CTCCAACACGACTTCATCCA-3' (forward), 5'-GACACAGTTCACCACTCCTA-3' (reverse) for TNFR2; 5'-ACCACAGTCCATGCATC AC-3' (forward), 5'-TCCACCACCCTGTTGCTGTA-3' (reverse) for GAPDH; and 5'-GAGTGACAAGCCTGTAGCCCATGTTGTAGCA-3' (forward), 5'-G CAATGATCCCAAAGTAGACCTGCCCAGAC-3' (reverse) for TNF-α. For α competitive RT-PCR, a known amount of a DNA competitor was added to the PCR reaction mixture. The DNA competitors were constructed using a DNA competitor kit (Takara), which was designed to be amplified by the same primers as the target cDNA, and which yielded a distinguishable size of PCR product. RT-PCR assays were performed three times and representative results are shown. GAPDH was used as an internal control.
Preparation of nuclear extracts
Human fetal astrocytes were stimulated with TNF-α (100 U/mL) for 30 min, and the nuclear extracts were prepared.30 Briefly, cells were harvested by scraping and then pelleted. Next the cells were resuspended in 1 mL of buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 10 µg/mL of leupeptin and aprotinin), and incubated on ice for 10 min. The cells were then lysed using 0.6% NP-40, and a nucleus pellet was obtained. The nucleus pellets were resuspended in 30 µL of hypertonic buffer C (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM DTT, 1 mM PMSF, 1 µg/mL of leupeptin and aprotinin) and the nuclear extracts were obtained. The concentration of nuclear protein was determined using the Bio-Rad protein assay.
Electrophoretic mobility shift assay (EMSA)
NF-κB activation was determined by electrophoretic mobility shift assay using the method described by Chaturvedi et al.31 Briefly, 8 µg of nuclear extracts were incubated for 15 min with or without a competitor in the binding buffer (10 mM Tris, pH 8.0, 0.075 mM KCl, 10% glycerol, 0.1 mM EDTA, 2.5 mM MgCl2, and 1 µg of poly dI/dC). 32P-end-labeled double stranded NF-κB binding oligonucleotide, 5'-AGTTGAGGGGACTT TCCCAGGC-3', was added to the reaction mixture and incubated on ice for an additional 30 min. The reaction mixtures were then resolved by electrophoresis. Dried gels were exposed to X-ray film (Kodak) at -70℃, and the specificity of binding was examined by competition with the unlabeled oligonucleotide.
Preparation of cellular extracts
To assess the intracellular protein level of TNFR1 and TNFR2, cellular extracts were prepared using a modification of the method of Dignam et al.30 After stimulation, the cells were washed with ice-cold phosphate buffered saline, harvested, and resuspended on ice in 300 µL of buffer A for 10 min. The cells were then lysed using buffer C containing 0.1% of NP-40, and the cellular extracts were obtained. The collected cellular extracts were stored at -70℃ until use, and protein concentrations were determined using a Bio-Rad protein assay.
Enzyme linked immunosorbent assay (ELISA)
Immunoreactive TNFR1 and TNFR2 protein levels were measured using TNF1 and TNFR2 Quantikine kits in accordance with the manufacturers' instructions (R&D Systems). All standards and samples were assayed in triplicate and the values were averaged. The assays were sensitive to TNFR1 and TNFR2 levels of > 7.8 pg/mL.
RESULTS
Steady state levels of TNFR1 and TNFR2 mRNA and protein in human fetal astrocytes
Steady state levels of TNFR1 and TNFR2 mRNAs were assessed by competitive RT-PCR on unstimulated astrocytes. The mRNA for TNFR1 was constitutively expressed at a higher level (equivalent to 1000 copies of competitor, Fig. 1) than the TNFR2 transcripts (equivalent to 10 copies of competitor, Fig. 1). In addition to the mRNA level, the levels of intracellular TNFR1 and TNFR2 proteins were also determined by ELISA, and it was found that TNFR1 protein was constitutively expressed on unstimulated astrocytes (127.3 ± 6.3 pg/mL) and that TNFR2 was expressed at barely detectable levels (< 7.8 pg/mL).
Expressions of TNFR1 and TNFR2 after treatment with TNF-α, IL-1β IFN-γ and LPS in human fetal astrocytes
To determine the effects of TNF-α, IL-1β IFN-γ and LPS on the relative levels of TNFR1 and TNFR2 mRNA transcription, each reagent was added to the culture media up to a final concentration of 100 U/mL. The cultures were incubated in the presence of reagents for 2, 4, 8, 12, 16, and 24 h at 37℃. Total RNA was then isolated from the cells and competitive RT-PCR was performed as described above. PCR products were subsequently quantified using a densitometer and the relative changes in TNFR2 specific mRNA were calculated after normalizing the data to the GAPDH mRNA levels. TNF-α, IL-1β, and IFN-γ induced an increase in TNFR2 mRNA in astrocytes, as shown by the competitive RT-PCR (Fig. 2). The increase in TNFR2 mRNA was evident at 2 h post-challenge, gradually increased thereafter, and remained elevated at 24 h. However, the relative level of TNFR2 mRNA in the astrocytes did not show any significant change after LPS treatment versus the untreated control culture. TNFR1 mRNA also did not demonstrate any significant change after TNF-α, IL-1β, IFN-γ, or LPS treatment (Fig. 2).
TNFR1 was constitutively shedded in unstimulated human astrocytes
Soluble TNFR1 and soluble TNFR2 were measured in the supernatant of human astrocyte cultures. Astrocytes were trypsinized and plated in a 24 well plate. After 24-48 hours the culture media was changed and the cells were harvested at 1, 6, 12, 24, 36, 48, 60, and 72 h. Soluble TNFR1 and sTNFR2 levels in the culture supernatant were determined using ELISA. The level of immunoreactive sTNFR1 increased steadily through the 72 hour period (1.5 ± 0.7 pg/mL → 265 ± 11.4 pg/mL, Fig. 3), whereas the amount of sTNFR2 did not show any significant change (< 4.5 ± 0.56 pg/mL, Fig. 3).
Blocking TNFR1 results in the neutralization of the effect of TNF-α on NF-κB activation
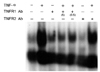
In addition to TNF receptor mRNA expression, we examined the functional roles of the receptors. To determine which receptor was responsible for NF-κB activation, we used neutralizing antibodies against TNFR1 and TNFR2. Cultures were incubated in the presence of antibodies for 1 h. After the removal of the media, new media containing 100U/mL of TNF-α was added. Thirty minutes later, nuclear extracts were harvested, and EMSA was performed using a 22 bp oligonucleotide. Nuclear extracts from the TNF-α stimulated cells showed a strong DNA-protein complex compared with the unstimulated astrocytes (Fig. 4). Neutralizing antibodies against TNFR1 decreased the level of the DNA-protein complex induced by TNF-α (Fig. 4). However, neutralizing antibodies against TNFR2 did not affect the complex formation (Fig. 4).
TNFR1 blocking results in the neutralization of the TNF-α effects on TNF-α induction in astrocytes
To determine which receptors consistently show major functioning, we examined the effects of neutralizing antibodies on TNF-α mRNA expression as induced by TNF-α. Cultures were treated with antibodies for 1 h and then stimulated with TNF-α for 2 h. After total RNA isolation, RT-PCR was performed. TNF-α mRNA was not observed in unstimulated astrocytes, but the astrocytes that were stimulated by TNF-α showed TNF-α mRNA expression. Neutralizing antibodies against TNFR 1 blocked the induction of TNF-α mRNA expression by TNF-α. However, neutralizing antibodies against TNFR2 did not affect this induction of TNF-α (Fig. 5).
DISCUSSION
The present experiment determined the relative mRNA levels and the protein levels of TNFR1 and TNFR2 in primary cultured human fetal astrocytes. From our RT-PCR studies, it is evident that both TNFR1 and TNFR2 mRNAs co-existe in human astrocytes. However, we found that TNFR1 mRNA is constitutively expressed at higher levels than the transcripts for TNFR2. Conflicting results have been reported regarding the expression of TNFR1 and TNFR2 in astrocytes. Dopp et al. reported that astrocytes expressed only TNFR1,32 whereas Lung et al. reported that TNFR1 and TFNR2 were co-expressed in rat astrocytes.27 Moreover, Tada et al. reported that TNFR1 and TNFR2 were co-expressed in human astrocytes.33 This discrepancy may be due to either the experimental method or the difference in species. We used competitive RT-PCR to quantify the mRNA expressions of TNFR1 and TNFR2. The obtained RT-PCR results demonstrated that TNFR1 mRNA was equivalent to 1000 copies of the TNFR1 competitor and that TNFR2 mRNA was equivalent to 10 copies of TNFR2 competitor in unstimulated human astrocytes. These results suggest that, although TNFR1 and TNFR2 mRNAs co-exist, TNFR1 mRNA is dominantly transcribed in human astrocytes. However, it is not clear that all of the TNFR1 and TNFR2 transcripts are translated into proteins in astrocytes. Therefore, we examined the intracellular protein levels of TNFR1 and TNFR2 by ELISA. As was the case for mRNA expression, relatively high levels of TNFR1 protein were detected (127.3 ± 6.3 pg/mL), whereas the levels of TNFR2 protein were undetectable (< 7.8 pg/mL). Surely, TNF receptor mRNA levels and intracellular protein levels may not correlate exactly with the surface TNF receptor. In fact, both TNFR1 and TNFR2 were very weakly detected on the cellular surfaces stained with immunofluorescence-labeled antibodies (data not shown). The difference between the intracellular level and the surface level of TNFR1 may arise from the intracellular remains of translated TNFR1 or from the extracellular shedding of membrane TNFR1.
Cellular activation by agents, such as LPS, and TNF-α, induces the rapid shedding of membrane TNFR in different types of cells by proteolytic cleavage of the cell surface receptors. As a result, soluble TNFR1 and sTNFR2 are generated.34,35
These soluble receptors have been shown to bind TNF-α with high affinity,36 and the shedding of TNF receptors may function as a potent inhibitor of the TNF-α.37,38 Despite the significant data concerning the function of sTNFRs as inhibitors of TNF-α activity, other reports show that when sTNFR concentrations are low, sTNFR may increase TNF-α activity by stabilizing the TNF-α trimeric structure and prolonging its availability for membrane receptor binding.39,40 We determined the levels of soluble TNFR1 (sTNFR1) and sTNFR2 in the astrocyte culture supernatant using ELISA. The level of sTNFR1 was found to increase gradually over a 72 h period, whereas sTNFR2 was not detectable. These results indicate that human astrocytes constitutively shed TNFR1 under normal conditions, which may be responsible for the weak expression of TNFR1 on the cellular surface. sTNFR1 is 5-30 times more potent than sTNFR2 in vitro and is also a better inhibitor of TNF-α activity in vivo.38,41,42 sTNFR1 secreted by astrocytes may participate in the control of TNF-α activity in the human brain. Further study concerning the physiologic role of sTNFR1 shed from astrocytes should be continues.
In the present study, we have shown that TNFR2 is selectively upregulated by IL-1β, TNF-α, or IFN-γ treatment in astrocytes, while LPS does not affect TNFR2 mRNA expression. In contrast, the TNFR1 transcript was found to maintain a constant level throughout the various cytokine treatments. The TNF receptor has been shown to be differentially regulated in various cell types, and similar modulation has been observed in epithelioid cells and in a fibroblast cell line for TNF-α and IL-1β.20 These results differ from those of a previous report, which found that TNF-α upregulates only TNFR1 in rat astrocytes and human epithelial cells.32 However, our results are consistent with the finding that TNF-α upregulates only TNFR2 in rat primary astrocytes.27 This difference may be attributed to the particular species or to the experimental method used. It is generally believed that TNFR1 mediates cytotoxicity, whereas TNFR2 mediates a mitogenic effect.12 Moreover, Selmaj et al. reported that both TNF-α and IL-6 act on astrocyte proliferation.24,43 Taken together with the current findings, it is possible that the proliferative effect of TNF-α on astrocytes might be mediated through the upregulation of TNFR2. Further studies should be undertaken to define the significance of TNFR2 upregulation by cytokines.
It has also been reported that exogenous TNF-α can induce TNF-α expression and NF-κB activation.10 Because TNFR2 has been shown to be involved in NF-κB activation,44 the fact that TNFR 1 is a predominantly expressed receptor on astrocytes does not necessarily prove that it is a the major signal transducing receptor for NF-κB activation. To determine which receptor is responsible for the NF-κB activation and the TNF-α expression, we used neutralizing antibodies against TNFR1 and TNFR2. The induction of TNF-α and NF-κB activation was inhibited using a neutralizing anti-TNFR1 antibody. These results suggest that the effects of TNF-α on NF-κB activation and TNF-α induction are mediated by TNFR1 in human astrocytes. In conclusion, the present study shows that TNFR1 is a predominantly transcribed and translated TNF receptor in human astrocytes and is constitutively shed from the cellular surface. Moreover, the study shows that TNFR1 functions in NF-κB activation and TNF-α induction. Although the expression of TNFR2 was at a very low level under normal conditions, it was increased by TNF-α, IL-1, or IFN-γ treatment. Further studies should be undertaken to define the significance of TNFR2 upregulation by cytokines and the physiologic role of the sTNFR1 secreted by human astrocytes.




 XML Download
XML Download