1. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999; 49:33–64. PMID:
10200776.

2. Zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000; 92:690–698. PMID:
10793105.
3. Clarke B, Chetty R. Cell cycle aberrations in the pathogenesis of squamous cell carcinoma of the uterine cervix. Gynecol Oncol. 2001; 82:238–246. PMID:
11531273.
4. Milde-Langosch K, Riethdorf S. Role of cell-cycle regulatory proteins in gynecological cancer. J Cell Physiol. 2003; 196:224–244. PMID:
12811815.
5. Funk JO. Cancer cell cycle control. Anticancer Res. 1999; 19:4772–4780. PMID:
10697591.
6. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999; 13:1501–1512. PMID:
10385618.
7. Semczuk A, Jakowicki JA. Alterations of pRb1-cyclin D1-cdk4/6-p16
INK4A pathway in endometrial carcinogenesis. Cancer Lett. 2004; 203:1–12. PMID:
14670612.
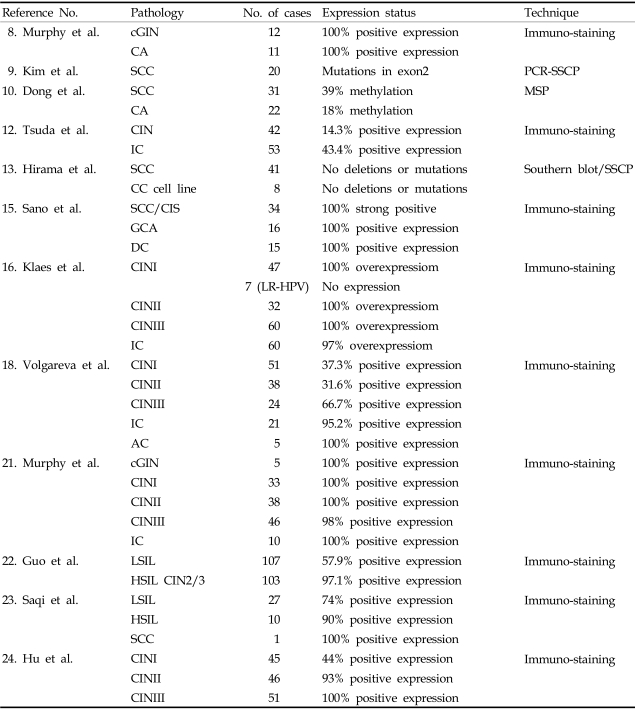
8. Murphy N, Heffron CC, King B, Ganuguapati UG, Ring M, McGuinness E, et al. p16
INK4A positivity in benign, premalignant and malignant cervical glandular lesions: a potential diagnostic problem. Virchows Arch. 2004; 445:610–615. PMID:
15378361.
9. Kim JR, Kim SY, Kim MJ, Kim JH. Alterations of CDKN2 (MTS1/p16INK4A) gene in paraffin-embedded tumor tissues of human stomach, lung, cervix and liver cancers. Exp Mol Med. 1998; 30:109–114. PMID:
9873831.
10. Dong SM, Kim HS, Rha SH, Sidransky D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001; 7:1982–1986. PMID:
11448914.
11. Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB, Herman JG. In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci USA. 1999; 96:12754–12759. PMID:
10535995.
12. Tsuda H, Hashiguchi Y, Nishimura S, Kawamura N, Inoue T, Yamamoto K. Relationship between HPV typing and abnormality of G1 cell cycle regulators in cervical neoplasm. Gynecol Oncol. 2003; 91:476–485. PMID:
14675665.
13. Hirama T, Miller CW, Wilczynski SP, Koeffler HP. p16 (CDKN2/cyclin-dependent kinase-4 inhibitor/multiple tumor suppressor-1) gene is not altered in uterine cervical carcinomas or cell lines. Mod Pathol. 1996; 9:26–31. PMID:
8821952.
14. Kelley MJ, Otterson GA, Kaye FJ, Popescu NC, Johnson BE, Dipaolo JA. CDKN2 in HPV-positive and HPV-negative cervical-carcinoma cell lines. Int J Cancer. 1995; 63:226–230. PMID:
7591209.
15. Sano T, Masuda N, Oyama T, Nakajima T. Overexpression of p16 and p14ARF is associated with human papillomavirus infection in cervical squamous cell carcinoma and dysplasia. Pathol Int. 2002; 52:375–383. PMID:
12100520.
16. Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, et al. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer. 2001; 92:276–284. PMID:
11291057.
17. Marjoniemi VM. Immunohistochemistry in gynaecological pathology: a review. Pathology. 2004; 36:109–119. PMID:
15203746.
18. Volgareva G, Zavalishina L, Andreeva Y, Frank G, Krutikova E, Golovina D, et al. Protein p16 as a marker of dysplastic and neoplastic alterations in cervical epithelial cells. BMC Cancer. 2004; 4:58. PMID:
15339339.
19. Giarre M, Caldeira S, Malanchi I, Ciccolini F, Leao MJ, Tommasino M. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle Arrest. J Virol. 2001; 75:4705–4712. PMID:
11312342.
20. Von Knebel Doeberitz M. New molecular tools for efficient screening of cervical cancer. Dis Markers. 2001; 17:123–128. PMID:
11790875.
21. Murphy N, Ring M, Killalea AG, Uhlmann V, O'Donovan M, Mulcahy F, et al. p16INK4A as a marker for cervical dyskaryosis: CIN and cGIN in cervical biopsies and ThinPrep smears. J Clin Pathol. 2003; 56:56–63. PMID:
12499437.
22. Guo M, Hu L, Baliga M, He Z, Hughson MD. The predictive value of p16(INK4a) and hybrid capture 2 human papillomavirus testing for high-grade cervical intraepithelial neoplasia. Am J Clin Pathol. 2004; 122:894–901. PMID:
15539381.
23. Saqi A, Pasha TL, McGrath CM, Yu GH, Zhang P, Gupta P. Overexpression of p16INK4A in liquid-based specimens (SurePath) as marker of cervical dysplasia and neoplasia. Diagn Cytopathol. 2002; 27:365–370. PMID:
12451568.
24. Hu L, Guo M, He Z, Thornton J, McDaniel LS, Hughson MD. Human papillomavirus genotyping and p16INK4a expression in cervical intraepithelial neoplasia of adolescents. Mod Pathol. 2005; 18:267–273. PMID:
15492761.
25. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004; 145:5439–5447. PMID:
15331580.
26. Reutens AT, Fu M, Wang C, Albanese C, McPhaul MJ, Sun Z, et al. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol Endocrinol. 2001; 15:797–811. PMID:
11328859.
27. Wang C, Pattabiraman N, Zhou JN, Fu M, Sakamaki T, Albanese C, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol Cell Biol. 2003; 23:6159–6173. PMID:
12917338.
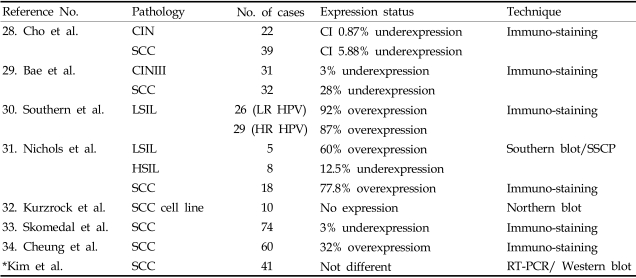
28. Cho NH, Kim YT, Kim JW. Correlation between G1 cyclins and HPV in the uterine cervix. Int J Gynecol Pathol. 1997; 16:339–347. PMID:
9421073.
29. Bae DS, Cho SB, Kim YJ, Whang JD, Song SY, Park CS, et al. Aberrant expression of cyclin D1 is associated with poor prognosis in early stage cervical cancer of the uterus. Gynecol Oncol. 2001; 81:341–347. PMID:
11371120.
30. Southern SA, Herrington CS. Differential cell cycle regulation by low- and high-risk human papillomaviruses in low-grade squamous intraepithelial lesions of the cervix. Cancer Res. 1998; 58:2941–2945. PMID:
9679950.
31. Nichols GE, Williams ME, Gaffey MJ, Stoler MH. Cyclin D1 gene expression in human cervical neoplasia. Mod Pathol. 1996; 9:418–425. PMID:
8729983.
32. Kurzrock R, Ku S, Talpaz M. Abnormalities in the PRAD1 (CYCLIN D1/BCL-1) oncogene are frequent in cervical and vulvar squamous cell carcinoma cell lines. Cancer. 1995; 75:584–590. PMID:
7812927.
33. Skomedal H, Kristensen GB, Lie AK, Holm R. Aberrant expression of the cell cycle associated proteins TP53, MDM2, p21, p27, CDK4, cyclin D1, RB, and EGFR in cervical carcinomas. Gynecol Oncol. 1999; 73:223–228. PMID:
10329038.
34. Cheung TH, Yu MM, Lo KW, Yim SF, Chung TK, Wong YF. Alteration of cyclin D1 and CDK4 gene in carcinoma of uterine cervix. Cancer Lett. 2001; 166:199–206. PMID:
11311493.
35. Psyrri A, DeFilippis RA, Edwards AP, Yates KE, Manuelidis L, DiMaio D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res. 2004; 64:3079–3086. PMID:
15126344.
36. Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004; 18:2699–2711. PMID:
15545627.
37. Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995; 11:211–219. PMID:
7624138.
38. Yoshinouchi M, Hongo A, Takamoto N, Ono Y, Nagao S, Miyagi Y, et al. Alteration of the CDKN2/p16 gene is not required for HPV positive uterine cervical cancer cell lines. Int J Oncol. 2000; 16:537–541. PMID:
10675486.
39. Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004; 430:226–231. PMID:
15241418.
40. Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-cdk protein kinase activity, is related to p21. Cell. 1994; 78:67–74. PMID:
8033213.
41. Hall M, Bates S, Peters G. Evidence for different modes of action of cyclin-dependent kinase inhibitors: p15 and p16 bind to kinases, p21 and p27 bind to cyclins. Oncogene. 1995; 11:1581–1588. PMID:
7478582.
42. Lukas J, Muller H, Bartkova J, Spitkovsky D, Kjerulff AA, Jansen-Durr P, et al. DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve the cell's requirement for cyclin D1 function in G1. J Cell Biol. 1994; 125:625–638. PMID:
8175885.
43. Salcedo M, Taja L, Utrera D, Chavez P, Hidalgo A, Perez C, et al. Changes in retinoblastoma gene expression during cervical cancer progression. Int J Exp Pathol. 2002; 83:275–286. PMID:
12657136.
44. Muller H, Lukas J, Schneider A, Warthoe P, Bartek J, Eilers M, et al. Cyclin D1 expression is regulated by the retinoblastoma protein. Proc Natl Acad Sci USA. 1994; 91:2945–2949. PMID:
8159685.
45. Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA. Physical interaction of the retinoblastoma protein with human D cyclins. Cell. 1993; 73:499–511. PMID:
8490963.
46. Sano T, Oyama T, Kashiwabara K, Fukuda T, Nakajima T. Immunohistochemical overexpression of p16 protein associated with intact retinoblastoma protein expression in cervical cancer and cervical intraepithelial neoplasia. Pathol Int. 1998; 48:580–585. PMID:
9736404.
47. Amortegui AJ, Meyer MP, Elborne VL, Amin RM. p53, retinoblastoma gene product, and cyclin protein expression in human papillomavirus virus DNA-positive cervical intraepithelial neoplasia and invasive cancer. Mod Pathol. 1995; 8:907–912. PMID:
8751330.
48. Choo KB, Chong KY. Absence of mutation in the p53 and the retinoblastoma susceptibility genes in primary cervical carcinomas. Virology. 1993; 193:1042–1046. PMID:
8384745.
49. Chetty R, Bramdev A, Aguirre-Arteta A, Pegoraro RJ, Sataar N. Relation between retinoblastoma and p53 proteins in human papilloma viruses 16/18 positive and negative cancers of the uterine cervix. J Clin Pathol. 1997; 50:413–416. PMID:
9215125.
50. Scheffner M, Munger K, Byrne JC, Howley PM. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci USA. 1991; 88:5523–5527. PMID:
1648218.
51. Fiedler M, Muller-Holzner E, Viertler HP, Widschwendter A, Laich A, Pfister G, et al. High level HPV-16 E7 oncoprotein expression correlates with reduced pRb-levels in cervical biopsies. FASEB J. 2004; 18:1120–1122. PMID:
15155561.
52. El-Deiry WS, Harper JW, O'Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994; 54:1169–1174. PMID:
8118801.
53. Michieli P, Chedid M, Lin D, Pierce JH, Mercer WE, Givol D. Induction of WAF1/CIP1 by a p53-independent pathway. Cancer Res. 1994; 54:3391–3395. PMID:
8012956.
54. Lie AK, Skarsvag S, Skomedal H, Haugen OA, Holm R. Expression of p53, MDM2, and p21 proteins in high-grade cervical intraepithelial neoplasia and relationship to human papillomavirus infection. Int J Gynecol Pathol. 1999; 18:5–11. PMID:
9891236.
55. Kim YT, Cho NH, Park SW, Kim JW. Underexpression of cyclin-dependent kinase (CDK) inhibitors in cervical carcinoma. Gynecol Oncol. 1998; 71:38–45. PMID:
9784316.
56. Huang LW, Chou YY, Chao SL, Chen TJ, Lee TT. p53 and p21 expression in precancerous lesions and carcinomas of the uterine cervix: overexpression of p53 predicts poor disease outcome. Gynecol Oncol. 2001; 83:348–354. PMID:
11606096.
57. Van de Putte G, Holm R, Lie AK, Trope CG, Kristensen GB. Corrigendum to "Expression of p27, p21, and p16 protein in early squamous cervical cancer and its relation to prognosis". Gynecol Oncol. 2004; 92:730.
58. Giannoudis A, Herrington CS. Differential expression of p53 and p21 in low grade cervical squamous intraepithelial lesions infected with low, intermediate, and high risk human papillomaviruses. Cancer. 2000; 89:1300–1307. PMID:
11002226.
59. Werness BA, Wang HQ, Chance J, Goldstein DJ. p53-independent expression of p21
WAF1/Cip1 in preinvasive and invasive squamous neoplasms of the uterine cervix. Mod Pathol. 1997; 10:578–584. PMID:
9195575.
60. Lu X, Toki T, Konishi I, Nikaido T, Fujii S. Expression of p21
WAF1/CIP1 in adenocarcinoma of the uterine cervix: a possible immunohistochemical marker of a favorable prognosis. Cancer. 1998; 82:2409–2417. PMID:
9635534.
61. Graflund M, Sorbe B, Karlsson M. Immunohistochemical expression of p53, bcl-2, and p21(WAF1/CIP1) in early cervical carcinoma: correlation with clinical outcome. Int J Gynecol Cancer. 2002; 12:290–298. PMID:
12060451.
62. Cheung TH, Lo KW, Yu MM, Yim SF, Poon CS, Chung TK, et al. Aberrant expression of p21(WAF1/CIP1) and p27(KIP1) in cervical carcinoma. Cancer Lett. 2001; 172:93–98. PMID:
11595134.
63. Sgambato A, Cittadini A, Faraglia B, Weinstein IB. Multiple functions of p27(KIP1) and its alterations in tumor cells: a review. J Cell Physiol. 2000; 183:18–27. PMID:
10699962.
64. Shiozawa T, Shiohara S, Kanai M, Konishi I, Fujii S, Nikaido T. Expression of the cell cycle regulator p27 (KIP1) in normal squamous epithelium, cervical intraepithelial neoplasia, and invasive squamous cell carcinoma of the uterine cervix. Immunohistochemistry and functional aspects of p27(KIP1). Cancer. 2001; 92:3005–3011. PMID:
11753978.
65. Huang LW, Chao SL, Hwang JL, Chou YY. Down-regulation of p27 is associated with malignant transformation and aggressive phenotype of cervical neoplasms. Gynecol Oncol. 2002; 85:524–528. PMID:
12051885.
66. Goff BA, Sallin J, Garcia R, VanBlaricom A, Paley PJ, Muntz HG. Evaluation of p27 in preinvasive and invasive malignancies of the cervix. Gynecol Oncol. 2003; 88:40–44. PMID:
12504625.
67. Kim YT, Choi EK, Cho NH, Ko JH, Yang WI, Kim JW, et al. Expression of cyclin E and p27
KIP1 in cervical carcinoma. Cancer Lett. 2000; 153:41–50. PMID:
10779628.
68. Troncone G, Vetrani A, de Rosa G, Gerbasio D, Palombini L. Cyclin dependent kinase inhibitor p27
KIP1 expression in normal and neoplastic cervical epithelium. J Clin Pathol. 1999; 52:880–887. PMID:
10711250.
69. Sgambato A, Zannoni GF, Faraglia B, Camerini A, Tarquini E, Spada D, et al. Decreased expression of the CDK inhibitor p27
KIP1 and increased oxidative DNA damage in the multistep process of cervical carcinogenesis. Gynecol Oncol. 2004; 92:776–783. PMID:
14984940.
70. Dellas A, Schultheiss E, Leivas MR, Moch H, Torhorst J. Association of p27
KIP1, cyclin E and c-myc expression with progression and prognosis in HPV-positive cervical neoplasms. Anticancer Res. 1998; 18:3991–3998. PMID:
9891436.
71. Erlandsson F, Wahlby C, Ekholm-Reed S, Hellstrom AC, Bengtsson E, Zetterberg A. Abnormal expression pattern of cyclin E in tumour cells. Int J Cancer. 2003; 104:369–375. PMID:
12569561.
72. Geng Y, Yu Q, Whoriskey W, Dick F, Tsai KY, Ford HL, et al. Expression of cyclins E1 and E2 during mouse development and in neoplasia. Proc Natl Acad Sci USA. 2001; 98:13138–13143. PMID:
11687642.
73. Dirks PB, Rutka JT. Current concepts in neuro-oncology: the cell cycle-a review. Neurosurgery. 1997; 40:1000–1015. PMID:
9149259.
74. Quade BJ, Park JJ, Crum CP, Sun D, Dutta A.
In vivo cyclin E expression as a marker for early cervical neoplasia. Mod Pathol. 1998; 11:1238–1246. PMID:
9872657.
75. Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen-Durr P. Inactivation of the CDK inhibitor p27
KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996; 13:2323–2330. PMID:
8957073.
76. Southern SA, McDicken IW, Herrington CS. Loss of cytokeratin 14 expression is related to human papillomavirus type and lesion grade in squamous intraepithelial lesions of the cervix. Hum Pathol. 2001; 32:1351–1355. PMID:
11774168.
77. Southern SA, McDicken IW, Herrington CS. Evidence for keratinocyte immortalization in high-grade squamous intraepithelial lesions of the cervix infected with high-risk human papillomaviruses. Lab Invest. 2000; 80:539–544. PMID:
10780670.
78. Keating JT, Cviko A, Riethdorf S, Riethdorf L, Quade BJ, Sun D, et al. Ki-67, cyclin E, and p16
INK4 are complimentary surrogate biomarkers for human papilloma virus-related cervical neoplasia. Am J Surg Pathol. 2001; 25:884–891. PMID:
11420459.
79. Kanai M, Shiozawa T, Xin L, Nikaido T, Fujii S. Immunohistochemical detection of sex steroid receptors, cyclins, and cyclin-dependent kinases in the normal and neoplastic squamous epithelia of the uterine cervix. Cancer. 1998; 82:1709–1719. PMID:
9576293.
80. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004; 23:2825–2837. PMID:
15077146.
81. Dulic V, Lees E, Reed SI. Association of human cyclin E with a periodic G1-S phase protein kinase. Science. 1992; 257:1958–1961. PMID:
1329201.
82. Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000; 2:E65–E67. PMID:
10783254.
83. Jones DL, Alani RM, Munger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997; 11:2101–2111. PMID:
9284049.
84. Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003; 3:233–245. PMID:
12676582.
85. Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002; 23:417–425. PMID:
12237154.
86. Kanao H, Enomoto T, Ueda Y, Fujita M, Nakashima R, Ueno Y, et al. Correlation between p14(ARF)/p16 (INK4A) expression and HPV infection in uterine cervical cancer. Cancer Lett. 2004; 213:31–37. PMID:
15312681.
87. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995; 83:993–1000. PMID:
8521522.
88. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998; 92:725–734. PMID:
9529249.
89. Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998; 17:5001–5014. PMID:
9724636.
90. Milde-Langosch K, Riethdorf S, Kraus-Poppinghaus A, Riethdorf L, Loning T. Expression of cyclin-dependent kinase inhibitors p16
MTS1, p21
WAF1, and p27
KIP1 in HPV-positive and HPV-negative cervical adenocarcinomas. Virchows Arch. 2001; 439:55–61. PMID:
11499840.
91. Brooks LA, Sullivan A, O'Nions J, Bell A, Dunne B, Tidy JA, et al. E7 proteins from oncogenic human papillomavirus types transactivate p73: role in cervical intraepithelial neoplasia. Br J Cancer. 2002; 86:263–268. PMID:
11870517.
92. Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997; 90:809–819. PMID:
9288759.
93. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992; 69:1237–1245. PMID:
1535557.
94. Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993; 13:4107–4114. PMID:
7686617.
95. Dellas A, Schultheiss E, Almendral AC, Gudat F, Oberholzer M, Feichter G, et al. Altered expression of mdm-2 and its association with p53 protein status, tumor-cell-proliferation rate and prognosis in cervical neoplasia. Int J Cancer. 1997; 74:421–425. PMID:
9291432.
96. Cheng YT, Li YL, Wu JD, Long SB, Tzai TS, Tzeng CC, et al. Overexpression of MDM-2 mRNA and mutation of the p53 tumor suppressor gene in bladder carcinoma cell lines. Mol Carcinog. 1995; 13:173–181. PMID:
7619220.
97. Skomedal H, Kristensen GB, Nesland JM, Borresen-Dale AL, Trope C, Holm R. TP53 alterations in relation to the cell cycle-associated proteins p21, cyclin D1, CDK4, RB, MDM2, and EGFR in cancers of the uterine corpus. J Pathol. 1999; 187:556–562. PMID:
10398121.
98. Ikenberg H, Matthay K, Schmitt B, Bauknecht T, Kiechle-Schwarz M, Goppinger A, et al. p53 mutation and MDM2 amplification are rare even in human papillomavirus-negative cervical carcinomas. Cancer. 1995; 76:57–66. PMID:
8630877.
99. Troncone G, Martinez JC, Palombini L, De Rosa G, Mugica C, Rodriguez JA, et al. Immunohistochemical expression of mdm2 and p21
WAF1 in invasive cervical cancer: correlation with p53 protein and high risk HPV infection. J Clin Pathol. 1998; 51:754–760. PMID:
10023338.
100. Oka K, Suzuki Y, Nakano T. Expression of p27 and p53 in cervical squamous cell carcinoma patients treated with radiotherapy alone: radiotherapeutic effect and prognosis. Cancer. 2000; 88:2766–2773. PMID:
10870059.
101. Busby-Earle RM, Steel CM, Williams AR, Cohen B, Bird CC. p53 mutations in cervical carcinogenesis-low frequency and lack of correlation with human papillomavirus status. Br J Cancer. 1994; 69:732–737. PMID:
8142262.
102. Milde-Langosch K, Albrecht K, Joram S, Schlechte H, Giessing M, Loning T. Presence and persistence of HPV infection and p53 mutation in cancer of the cervix uteri and the vulva. Int J Cancer. 1995; 63:639–645. PMID:
7591279.
103. Horner SM, DeFilippis RA, Manuelidis L, DiMaio D. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J Virol. 2004; 78:4063–4073. PMID:
15047823.
104. Helland A, Holm R, Kristensen G, Kaern J, Karlsen F, Trope C, et al. Genetic alterations of the TP53 gene, p53 protein expression and HPV infection in primary cervical carcinomas. J Pathol. 1993; 171:105–114. PMID:
8283348.
105. Benjamin I, Saigo P, Finstad C, Takahashi H, Federici M, Rubin SC, et al. Expression and mutational analysis of p53 in stage IB and IIA cervical cancers. Am J Obstet Gynecol. 1996; 175:1266–1271. PMID:
8942499.
106. Avall-Lundqvist EH, Silfversward C, Aspenblad U, Nilsson BR, Auer GU. The impact of tumour angiogenesis, p53 overexpression and proliferative activity (MIB-1) on survival in squamous cervical carcinoma. Eur J Cancer. 1997; 33:1799–1804. PMID:
9470836.
107. Cho NH, Lim SY, Kim YT, Kim D, Kim YS, Kim JW. G2 checkpoint in uterine cervical cancer with HPV 16 E6 according to p53 polymorphism and its screening value. Gynecol Oncol. 2003; 90:15–22. PMID:
12821336.
108. Nurse P. Ordering of S phase and M phase in the cell cycle. Cell. 1994; 79:547–550. PMID:
7954820.
109. Zehbe I, Ratsch A, Alunni-Fabbroni M, Burzlaff A, Bakos E, Durst M, et al. Overriding of cyclin-dependent kinase inhibitors by high and low risk human papillomavirus types: evidence for an
in vivo role in cervical lesions. Oncogene. 1999; 18:2201–2211. PMID:
10327066.
110. Theron T, Bohm L. Influence of the G2 cell cycle block abrogator pentoxifylline on the expression and subcellular location of cyclin B1 and p34cdc2 in HeLa cervical carcinoma cells. Cell Prolif. 2000; 33:39–50. PMID:
10741643.
111. Hashiguchi Y, Tsuda H, Nishimura S, Inoue T, Kawamura N, Yamamoto K. Relationship between HPV typing and the status of G2 cell cycle regulators in cervical neoplasia. Oncol Rep. 2004; 12:587–591. PMID:
15289842.


 PDF
PDF ePub
ePub Citation
Citation Print
Print



 XML Download
XML Download