

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The disease designated "r(21)" is a rare chromosomal anomaly characterized by deletions of part of chromosome 21, and is often associated with mental retardation and dysmorphic features.1 On the other hand, trisomy 21 is fairly common, occurring in approximately one out of every 700 births. To our knowledge, however, there has never been a report detailing the concurrence of r(21) and trisomy 21 in the children of normal parents. Here, we present the first case of two children of normal parents with two different abnormalities of chromosome 21, r(21)/idic(21) mosaicism and trisomy 21. Additionally, we include a review of relevant literature.
CASE REPORT
The patient was a ten-year-old girl who was admitted to our out-patient psychiatry department for observation to determine the extent and origin of her mental retardation. She was the second child of healthy, unrelated parents (mother aged 31, father 35 at her birth) and was delivered by Cesarean section, with a birth weight of three kg. She had no history of seizures, nor head trauma. She was thin, whereas her elder sister appeared well-nourished. She was 140 cm tall and weighed approximately 24 kg. She appeared to be somewhat nervous about being examined, was very hyperactive, easily distracted, and continuously nagged her mother with repeated and frivolous questions. Her mental age was assumed to be somewhere below the level of a normal three-year old. Her Social Quotient (SQ) was found to be 24. A battery of other tests confirmed that the girl was severely mental retarded according to DSM-IV criteria. She exhibited no anemia, jaundice, skin eruptions, purpura, lymphadenopathy, or hepatosplenomegaly. Peripheral blood counts showed a white cell count of 8.3×109/l with 42.4% neutrophils, 50.4% lymphocytes, 5.7% monocytes, 1.3% eosinophils, and 0.2% basophils. The red cell count was 5,100×109/l, hemoglobin was 14 g/dl, and platelet count was 300×109/l. Blood chemistry indicated normal liver and kidney function. Serum immunoglobulin concentrations were documented as follows: IgG 15.2 g/dl (normal range 7.0-16.0), IgA 2.5 g/dl (0.7-4.0), and IgM 1.6 g/dl (0.4-2.3). Results of a surface marker analysis of peripheral mononuclear cells were within normal limits and were as follows: 71% T cells, 12% B cells, and a CD4/CD8 ratio of 1.68.
Her elder sister also had mental retardation. She exhibited the physical characteristics associated with Down's syndrome: slanted eyes, epicanthic folds, and a flat nose. Her full-scale intelligence quotient (FSIQ) was below 40, indicating a moderate degree of mental retardation. However, the girls' parents were found to be both phenotypically and cytogenetically normal.
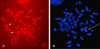
Cytogenetic analysis of peripheral lymphocytes with GTG banding revealed that the patient had a karyotype of 46,XX,r(21)(p13q22.3)[91]/46,XX, idic(21)(p13)[4]/46,XX[5] (Fig. 1A, B). The breakpoint of r(21) was located at q22.3. Her elder sister had a karyotype of 46,XX,rob(14;21)(q10;q10),+21 (Fig. 1C). C-banding of the patient's chromosomes revealed that r(21) was monocentric and i(21) was dicentric (Fig. 1D). The r(21) of the patient was positive for Ag-NOR staining, consistent with the satellite region from the intermediate i(21) via asymmetrical breakage; however, that of her mother was not informative (data not shown). FISH, performed using the AneuCyte™ 21 (21q22.2) unique sequence probe (Cytocell Ltd., Banbury, Oxfordshire, UK) and 21 telomere-specific probe (Q-biogene, Inc., Carlsbad, CA, USA), identified r(21) and i(21) each from different fields of the patient (Fig. 2A, B), and showed positive signals on the telomeric region of the maternal 21q (data not shown). Molecular studies using polymorphic markers (D7S820, D8S1179, D18S51, and D21S11) located on various chromosomes revealed that both children were of the same parentage and that the elder sister's extra chromosome 21 was of maternal origin (Fig. 3A). All analyses were performed on a 3100 ABI Prism Genetic Analyzer™ (Applied Biosystems), according to the manufacturer's instructions. Data were processed by GeneScan™ and Genotyper™ software (Applied Biosystems). A maternal origin was determined for the r(21) of the patient, on the basis of DNA polymorphisms at the D21S1411 and D21S1446 loci (Fig. 3B).
DISCUSSION
The sisters had different abnormalities involving chromosome 21, rob(14q21q) and r(21)/idic (21) mosaicism. Acrocentric rearrangements are involved in 5% of all Down syndrome cases and 95% of these involve Robertsonian translocations.2 About half of rob(14q21q) are inherited; however, over 95% of rea(21q21q) arise de novo.3 Because the parents were cytogenetically and phenotypically normal, the siblings' chromosome 21 abnormalities were considered to be formed de novo.
In general, Down's syndrome patients are menmentally retarded, but are relatively able to function socially. The mentality and sociality of the patient were markedly more severely retarded than those of her sister, who had partial trisomy of chromosome 21. There exists a wide range of phenotypic variation among r(21) carriers, which can be largely attributed to different breakpoints and chromosomal instability. This range includes severely affected individuals who exhibit clinical manifestations of 21q- syndrome,2 almost-normal individuals,4,5 and phenotypically abnormal patients exhibiting features of Down's syndrome.6,7 Also, individuals with similar phenotypes have similar mitotic stability and breakpoints within the almost-normal group.8 The r(21) of the patient comprised 91% of her lymphocytes, indicating that the r(21) found in this patient is mitotically stable in the lymphocytes.
Three mechanisms have been suggested for the formation of the r(21) chromosome.9 In the first case, a monocentric r(21) chromosome may be formed by the breakage of both arms and the reunion of the chromosome breakpoints. In the second case, a double-sized symmetric dicentric r(21) chromosome could be formed by an exchange of sister chromatids, occurring in a monocentric r(21) chromosome. Another suggested mechanism involves the generation of an asymmetric dicentric r(21) chromosome via the asymmetric breakage and reunion of the long arms of an intermediate dicentric isochromosome, resulting from de novo 21p;21p translocation.10 In another case report, a U-type exchange took place during maternal meiosis between the distal 21q regions on both arms of the der(21) chromosome and was associated with the distal 21q duplication. This was suggested as the third possible mechanism for the formation of a monocentric ring chromosome.11 In our patient, however, the monocentric r(21) was formed de novo and her mother was cytogenetically normal. Because the ring lacks a 21qter segment and the isochromosome does not , in FISH, the isochromosome must have preceded the ring. Therefore, a monocentric r(21) of our patient may be generated via the asymmetric breakage and reunion of the breakpoints of an intermediate dicentric isochromosome, resulting from de novo 21p;21p translocation.
To our knowledge, there have been no reports describing a case of concurrence of r(21)/idic(21) mosaicism and trisomy 21 in full sisters of normal parents, as above. Molecular analysis using polymorphic markers revealed that the same maternal chromosome 21 involved in rob(14q21q) was shown to have formed the r(21). We tried to find some cryptic abnormalities on the maternal chromosome 21 using a FISH probe, C-banding, and Ag-NOR staining. The intact long arm was obtained, but did not reveal any informative result about the short arm. Generally, it is known that the majority of de novo rob(14q21q) are formed during maternal meiosis and the parental origin of the de novo isochromosomes are roughly equally divided between maternally and paternally derived rearrangements.12 We suspect that some cryptic abnormalities on the maternal 21p were the source of the children's abnormalities, but further evaluation is necessary.


 XML Download
XML Download