

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The retraction of the podocyte foot processes into their cell bodies and spacing-out of the filtration slits constitute the hallmark ultrastructural changes seen in the minimal change nephrotic syndrome for humans and for the corresponding rat models.1-3 Proteinuria associated with glomerular diseases is secondary to alterations of the charge-selective and/or size-selective properties of the GBM, but molecular modifications that are responsible for these functional changes are still poorly understood.4 Although the role of the glomerular basement membrane (GBM) for restricting the filtration of macromolecules has been emphasized for nearly two decades,1,5 several recent studies have shown that slit diaphragms located between the foot processes may play a critical role as barriers to retain macromolecules.6-10 Pavenstadt et al.11 proposed several physiologic functions of podocyte: First, they function as a specific pericyte counteracting the high transmural distending forces to permit the high-pressure perfusion of glomerular capillaries. Second, podocytes are crucially involved in establishing the specific permeability properties of the glomerular filter. Third, podocytes are responsible for the continuous cleaning of the filter. Yet there was no direct evidence offered for supporting these hypotheses. Puromycin aminonucleoside (PAN)-induced nephrosis is a well-described model of human idiopathic nephrotic syndrome, but the mechanism of PAN's effect is not completely understood. Smithies12 has recently emphasized that the single nephron glomerular filtration rate (GFR) is a prime factor in determining the development of proteinuria. He thought that severe pathological decreases in the slit diaphragm length seen in minimal-change nephrotic syndrome for humans and for animals treated with puromycin aminonucleoside, or for humans or animals with mutations in the gene coding for nephrin, can cause albuminuria by the reduction of the single nephron GFR.
In recent years, several molecules have been reported to be associated with the slit diaphragm13-16. Zonula occludens-1 (ZO-1), which is a protein found on the cytoplasmic face of tight junctions, is also expressed on the cytoplasmic surface of podocyte foot processes at the point of insertion of the slit diaphragm13. Several reports showed that the change of ZO-1 distribution and/or its expression in podocytes is related with proteinuria.10,17,18 Kawachi's experiments10 demonstrated that monoclonal antibody 5-1-6 alters the expression of both nephrin and ZO-1 proteins in rat podocytes. Although Kurihara et al.18 described the altered ZO-1 protein distribution in podocytes of a PAN treated rat model, they did not show the quantitative change of ZO-1 protein expression.
The antiproteinuric effect of cyclosporin A (CsA) has been reported in several human and animal studies. In both children and adults,19-21 CsA is an option for those who have not responded to conventional steroid treatment. The pharmacological antiproteinuric effect of CsA has long been demonstrated both experimentally and clinically. Meyrier22 already stressed that the mode of action of CsA in reducing or suppressing proteinuria in glomerular diseases is not merely linked to its immunosuppressive virtues. In fact, several lines of evidence, both from experimental evidence and in human studies, have led to the belief that CsA exerts a non-immunologic, antiproteinuric effect of its own. Jameson et al.23 have demonstrated a marked effect of CsA on the glomerular filtration barrier that was independent of renal perfusion in a study of filtration by glomeruli isolated from Wistar-Kyoto rats. Zietse et al.24 have shown that CsA induced complete or partial remissions in patients with various nephrotic syndromes, with no consistent change being noted for the GFR. Kokui et al.25 have reported that CsA can reduce proteinuria for rats with PAN nephrosis, but CsA cannot ameliorate the glomerular changes seen upon examination with a conventional light microscope. Recently, Luimula et al.26 have shown an alteration of protein levels for several podocyte associated molecules in PAN induced nephrosis of rats, and these alterations seem to be associated with the development of a proteinuria, but there has been no report on the effect of CsA on the podocyte-associated molecules in PAN nephrosis.
In this study, we have adopted a PAN-induced rat model nephrosis that represents a minimal change nephrotic syndrome in humans, and we investigated whether proteinuria is associated with alteration of ZO-1 expression within the glomeruli, and whether CsA diminished proteinuria and ZO-1 protein expression.
MATERIALS AND METHODS
Experimental group models
Eighteen male Sprague-Dawley rats (150 to 200 g) were used in this study. The animals were treated according to the rules and regulations of the Ethical Committee of the Yonsei University. The rats were fed standard rat chow and they had a free access to tap water. Twelve rats received a single intraperitoneal injection of PAN (15 mg/100 g; Sigma-Aldrich Co., St. Louis, MO, U.S.A.), which was dissolved in saline at a dilution of 20 mg/ml. The other six rats received an equal volume of saline (normal control group; control). From the day of PAN injection (Day 0), CsA solution for intravenous use (Chongkundang Pharmaceutical Corp., Seoul, Korea) was diluted 1 : 10 with saline and then administered intraperitoneally once a day for 20 days (n=6, PAN+CsA). The remaining six rats received PAN, but they didn't receive CsA (n=6, nephrotic control rats; PAN).
Urine was collected overnight from the metabolic cages at days 0, 9 and 18 after the day of the PAN injection. The urinary protein was measured with a Bio-Rad protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and by using a spectrophotometer. Serum creatinine was measured at 0 and 18 days after the PAN injection. At the end of the study (20 days after the PAN administration), the animals were sacrificed and their kidneys were removed and then processed for a Western blot procedure and immunohistchemistry.
Western blot analysis
Rat glomeruli were isolated by a sieving technique, and they were then lyzed in a sodium dodecyl sulfate (SDS) sample buffer [2% SDS, 10 mmol/L Tris-HCl, pH 6.8, 10% (vol/vol) glycerol]. The lysate mixture was centrifuged at 10,000 ×g for 10 minutes at 4℃, and the resultant supernatant was stored at 70℃. Protein concentrations were determined with a Bio-Rad kit (Bio-Rad). Aliquots of 50 µg protein were treated with Laemmli sample buffer, then heated at 95℃ for five minutes and finally they were electrophoresed at 50 µg/lane in a 6% acrylamide denaturing SDS-polyacrylamide gel. The proteins were next transferred to a Hybond-ECL membrane (Amersham Life Science, Inc., Arlington Heights, IL, U.S.A.) using a Bio-Rad semidry blotting apparatus (Bio-Rad). The membrane was incubated in a blocking buffer A [phosphate-buffered saline (PBS), 0.1% Tween-20, and 5% nonfat milk] for one hour at room temperature and then incubated overnight at 4℃ with a rabbit polyclonal antibody that was specifically reactive to the rat ZO-1. The membrane was washed once for 15 minutes and twice for five minutes in PBS with 0.1% Tween-20, and then the membrane was incubated in buffer A with horseradish peroxidase-linked sheep anti-mouse IgG (Amersham) at a 1 : 1000 dilution. The washes were repeated, and at last, the membrane was developed with a chemiluminescent agent (ECL; Amersham Life Science, Inc.).
Tissue processing and staining procedure
The renal tissue was fixed in 10% neutral-buffered formalin and then processed in the standard manner; 5 µm sections were utilized. The slides were deparaffinized, hydrated in ethyl alcohol and then washed in tap water. Antigen retrieval was carried out in a 10 mmol/L sodium citrate buffer for 20 minutes using a Black and Decker vegetable steamer. For ZO-1 staining, a polyclonal rabbit anti-rat ZO-1 antibody (Zymed Laboratories, Inc., San Francisco, CA, U.S.A.) diluted to 1:200 with 2% casein in BSA was applied for an overnight incubation at room temperature.
After washing, the biotinylated link anti-rabbit IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, U.S.A.) was added for 20 minutes. The slides were washed and then incubated with a streptoavidin-HRP (Santa Cruz) for 30 minutes. Diaminobenzidine was added for one minute and the slides were counterstained with hematoxylin.
The assessment of glomerular ZO-1 immunohistochemical staining was scored on a semiquantitative scale of 0 to 4 according to staining intensity that was compensated with the background staning intensity. All glomeruli in each section were examined by a pathologist blinded to the identity of the groups, and a staining score was obtained by multiplying the intensity of podocytes staining by the percentage of glomeruli staining for that intensity, these numbers were than added for each experimental animal to give the staining score. ∑(intensity of staining) × (% of glomeruli with that intensity)=staining score.
Statistical analysis
All values are expressed as the mean ± SEM. Statistical analysis was performed using the statistical package SPSS for Windows Version 11.0 (SPSS, Inc., Chicago, IL, USA). The results were analyzed using a Student's t-test or the Kruskal-Wallis nonparametric test for multiple comparisons.
RESULTS
Throughout the experiment, the rats' food and water intake was similar for all the treatment groups. Body weight was significantly higher for the control rats than for the other groups at day 20 (p<0.05 vs. PAN, PAN+CsA). Serum creatinine levels for the PAN-administered animals were significantly higher than those for the normal control animals (0.46 ± 0.08 mg/dL vs. 0.40 ± 0.07 mg/dL, p<0.01). Serum creatinine levels were significantly higher in the PAN+CsA group than in the PAN group on day 18 (0.55 ± 0.10, p<0.01) (Table 1).
Proteinuria
The normal control values for 24-hour urinary protein in the rats used in this study did not exceed 30 mg. Fig. 1 shows the 24-hour urinary protein values for the experimental rats following the injection of PAN. There was a gradual increase of proteinuria for the PAN rats (n=6) as compared to the control rats (n=6). This increase was ameliorated by the CsA treatment. Eighteen days after PAN administration, the mean value of 24-hour urinary protein excretion rose to 1021.9 ± 128.9 mg/day for the PAN group (p<0.01 vs. control). The CsA treatment induced a significant reduction in the proteinuria levels (556.4 ± 102.3 mg/day, p<0.01 vs. PAN).
Western blot
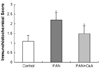
Fig. 2 shows the ZO-1 protein expression as estimated by the Western blot method in the control, PAN and PAN+CsA groups. Relying on information gleaned from previous research reports, the 225-kD band was selected and quantified for a statistical analysis.18 We found that there was a significant increase of ZO-1 proyein expression in PAN group (176%, p<0.01), and this increase was ameliorated by the CsA treatment (71.1%, p<0.05).
DISCUSSION
Passage of plasma proteins across the glomerular basement membrane is believed to be normally restricted by a charge-selective barrier and a size-selective barrier.27,28 although much of investigations have revealed that the defect in minimal-change glomerulopathy results mainly from a loss of charge selectivity,29 several recent studies have shown that slit diaphragms located between the foot processes may play a critical role as barriers to retain macromolecules.6-10
This study showed that the proteinuria induced by PAN was reduced by the CsA treatment, and these changes were accompanied by the change of ZO-1 protein expression in the glomeruli. PAN induced nephrosis is a well-described model of human idiopathic nephrotic syndrome30 and the glomerular morphologic changes seen in rats with PAN nephrosis closely resemble those in human minimal change nephrotic syndrome (MCNS). In addition, PAN targets the podocytes, inducing a focal segmental glomerulosclerosis (FSGS).31 However, the precise mechanisms of this PAN-induced proteinuria are not well understood. In the normal rat, the foot processes of podocytes are kept wide open to facilitate the passage of the glomerular filtrate, and they are held together by tenuous slit diaphragms that bridge the filtration slits. In contrast, in the PAN nephrosis rat, the foot processes are reduced in number, the filtration slits are greatly narrowed and the slit diaphragms are displaced by occluding-type junctions.18 The major morphologic changes in PAN nephrosis are seen as the effacement of the foot processes of glomerular visceral epithelial cells, or the podocytes.30,32,33 The podocyte is a complex cell with many remarkable structural and functional properties. As PAN treatment leads to profound changes in the well-organized cytoskeleton of podocytes,30 this process warrants a detailed molecular analysis of the diseased podocytes. Recent studies have shown remarkable changes both in nephrin and in the proteins directly associating with it.26 ZO-1 protein is a component of the slit diaphragm that seems to play a pivotal role in maintaining the permeable-selective properties of the glomerular capillary wall.18 ZO-1 protein has been reported in several studies as one of the proteins that may be associated with the development of proteinuria.10,17,18 Reiser et al.15 have demonstrated that the slit diaphragm represents an adherens junction that is composed of P-cadherin, ZO-1 and catenin.
Here in our study, we observed an up-regulation of ZO-1 protein in PAN nephrosis by the use of a Western blot technique and subsequent immunohistochemistry. From the immunohistochemical findings, we found that ZO-1 expression increased along the capillary wall side. Yet in our current study, we could not clarify the precise role of increased ZO-1, but it may be due to an adaptive response of the podocytes to maintain the filtration barrier during the PAN challenge. One possible explanation for this result is that ZO-1 protein might contribute in creating newly formed cell-to-cell junctions between the podocytes as a compensatory mechanism. Although Kurihara et al.18 showed an altered distribution of ZO-1 protein in PAN nephrosis, they did not demonstrate the quantitative analysis of ZO-1 protein. One major difference between our study and the previous one was the PAN treated time18. We treated PAN for almost 3 weeks, but previous data were obtained in much shorter experimental time. It may be possible that ZO-1 change in PAN treated glomeruli varies with time course.
Effacement of the foot processes, which is associated with proteinuria in many renal diseases, is reparable when appropriate therapeutic approaches are successfully employed. However, the specific regulators for this regeneration process are also unknown. Although recent studies have suggested the importance of the proteins constituting the slit diaphragm in developing proteinuria,26,34 there has been no data about the effects of CsA on the slit diaphragm associated protein in the nephrotic models. We found that CsA diminished both the ZO-1 protein expression of the glomeruli and the proteinuria in PAN nephrosis. The previous clinical studies have suggested that CsA may have a beneficial effect in reducing proteinuria in MCNS and the nephrotic syndrome that is associated with FSGS.35-39 The precise mechanism by which CsA reduces proteinuria in nephrotic syndrome and PAN nephrosis is largely unknown. Bustos et al.40 have shown that CsA modulates the glomerular production of inflammatory mediators in the PAN model. Wangs et al.41 reported that CsA helped to recover the diminished level of glomerular superoxide dismutase and also the level of catalase in PAN nephrosis. The amelioration of ZO-1 changes shown in the current study suggested that CsA had an effect on the proteins that help to make up the slit diaphragm, and so CsA might decrease proteinuria in the nephrotic models. But it is somewhat unclear whether the change of ZO-1 protein has any direct relationship with proteinuria or not. CsA usually takes effect by an immunologic mechanism, but several reports have demonstrated the effect of CsA on non-immunologic diseases such as Alport's syndrome and diabetic glomerulosclerosis.42,43 Jameson et al.23 studied filtration by subcutaneously injecting isolated rat glomeruli with either saline or CsA at a dose of 30 mg/kg/day for 2 - 3 weeks, and they demonstrated a pharmacologic effect of CsA on the glomerular filtration barrier in an experimental situation where no hemodynamic phenomenon could be at work. In a passive model of anti-GBM nephritis in the mouse, Berden et al.44 compared the effect of CsA on proteinuria and on GFR with that of a solvent. CsA was noted by Berdan and colleagues to reduced protein excretion and the GFR. To dissociate the effect of CsA on glomerular permeability to the proteins and the hemodynamic effect of the drug, the experiment was repeated before and after with the addition of phenoxybenzamine, a solvent which abolished glomerular vasoconstriction. Although both experimental conditions had similar GFRs, the albuminuria was significantly less with using CsA. Berden et al concluded that the antiproteinuric effect of CsA was essentially due to the reduced glomerular basement membrane permeability to albumin, and it was not due to a hemodynamic phenomenon. Zietse et al.24 studied 20 patients with nephrotic syndrome, and CsA induced complete remission in the patients with MCNS, with no consistent change being noted for the GFR. In the six patients with idiopathic membranous glomerulonephropathy (IMGN), CsA induced partial remission and the GFR was unchanged. Infusion of dextrans showed a decreased clearance for molecules of 50 - 58 A. Conversely, membranoproliferative glomerulonephritis (MPGN) and FSGS proteinuria did not respond to CsA despite a fall in GFR and decrease of a renal plasma flow of 20 to 33% respectively. The authors concluded that in MCNS, CsA had decreased the proteinuria through an increase of charge selectivity. In IMGN, CsA seemed to decrease proteinuria by an increase of pore size selectivity. So, the possible mechanism of CsA on PAN nephrosis could take place both ways. Further studies should be considered to unveil the precise mechanism of CsA on PAN nephrosis.
In conclusion, CsA treatment significantly reduced proteinuria and diminished the glomerular ZO-1 expression in a rat model PAN nephrosis. These findings suggest the potential role of slit diaphragm associated proteins in the development of the nephrotic syndrome, and CsA decreased the proteinuria probably by a direct action on the expression of these proteins in the podocyte. Further investigations are needed to clarify the role of slit diaphragm associated proteins in the development of PAN nephrosis.




 XML Download
XML Download