

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Soft-tissue sarcomas (STSs) are rare mesenchymal neoplasms accounting for approximately 1% of all adult malignancies (12). The classification of STS and approach to treatment has evolved in recent years with increasing understanding of the molecular pathogenesis and complex cytogenetics of these tumors. Conventional histology and immunohistochemistry still remain the cornerstones for diagnosis, with cytogenetics being increasingly incorporated into the diagnostic work up. Similarly, treatment of advanced STS is shifting from generic “one-fits-all” 106cytotoxic chemotherapy to histology-driven therapy aimed at targeting specific mutations and signaling pathways unique to that subtype of sarcoma (34). The earliest and the most successful example was the discovery of kit mutations in gastrointestinal stromal tumor (GIST), and subsequent development of the selective tyrosine kinase inhibitor imatinib, which revolutionized the treatment of GIST (3). The role of imaging in GISTs and response assessment is elaborated in a separate article in this issue given its significance.
In this article, we will provide a brief overview of the revised World Health Organization (WHO) classification of soft tissue tumors, discuss in detail the radiology and management of the two most common adult non-GIST STS, namely liposarcoma and leiomyosarcoma, and review some of the emerging histology-driven targeted therapies in non-GIST STS, focusing on the role of the radiologist.
Revised 2013 WHO Classification of STS
In 2013, based on the new immunohistochemical and cytogenetic data available, the WHO published its revised soft tissue tumor classification (5). As per the revised classification, depending on their biological behavior, soft tissue tumors are classified as benign (usually do not recur), intermediate-locally aggressive (often recur but do not metastasize), intermediate-rarely metastasizing (often recur and may metastasize in < 2% cases), or malignant (most common and have a high risk of metastases). Examples of these four are lipoma, well-differentiated liposarcoma, solitary fibrous tumor, and dedifferentiated liposarcoma, respectively (67).
The emphasis in the revised classification is on cytogenetics and molecular pathways, indicating the potential for treatment by molecular targeted therapies (MTTs) (67). Table 1 lists the molecular mechanisms for sarcomagenesis in a few important sarcomas. Based on cytogenetics, sarcomas tend to fall in either of two major subsets. One group is cytogenetically simple with a few chromosomal rearrangements leading to the formation of fusion proteins (891011). Examples of sarcomas in this group include synovial sarcoma with the t(X; 18) translocation resulting in SYT-SSX1 or SYT-SSX2 fusion protein (512) and Ewing sarcoma family of tumors with the classic t(11; 22) translocation forming EWS-FL1 fusion transcript, which acts as a c-myc pathway activator (1314). The other group includes sarcomas with complex karyotypes and multiple chromosomal abnormalities such as losses and gains, unbalanced translocations, and lack of fusion proteins (4101115). Examples of these include dedifferentiated liposarcoma, leiomyosarcoma, and pleomorphic sarcoma. Eventually, both these groups cause dysregulation of terminal signal pathways such as the vascular endothelial growth factor (VEGF), insulin-like growth factor 1, platelet-derived growth factor (PDGF), C-kit, phosphatidylinositol 3-kinase (PI3)/AKT/mechanistic target of rapamycin (mTor), and c-Met pathways (4915). All these factors are potential targets of MTTs (Fig. 1). Similar pathways are also responsible for carcinogenesis, and as such, many MTTs currently being used against carcinomas are also now increasingly used to treat sarcomas.
The revised classification and treatment options can have important implications for the radiologist especially from the aspect of treatment response. The increased use of MTTs has led to different manifestations of treatment response beyond the traditional size criteria, with tumor morphology and density becoming important aspects as well for the radiologist to factor in (16). Apart from the size-based Response Evaluation Criteria in Solid Tumors (RECIST), the Choi, mass, attenuation, size, and structure (MASS), and immune related response criteria have been introduced, amongst others, ushering in the era of personalized medicine and personalized radiology (16). Similarly, several new class-specific and drug-specific toxicities have been recognized (34).
Based on the tissue that they histologically resemble, STS are divided into 12 different categories (Table 2) (5). Most of the categories are relatively self-explanatory; for example, liposarcoma, leiomyosarcoma, and angiosarcoma show adipocytic, smooth muscle, and vascular differentiation, respectively. While discussing each category in detail is beyond the scope of this article, the salient changes compared to the 2002 classification are the inclusion of GISTs and peripheral nerve sheath tumors for the first time under STS, and the introduction of a new category of undifferentiated/unclassified tumors (167). The category ‘tumors of uncertain differentiation’ includes a heterogeneous subset of sarcomas whose tissue of origin is yet not clear; it includes tumors often seen in young adults such as synovial sarcoma, clear cell sarcoma, alveolar soft part sarcoma, extraskeletal Ewing sarcoma, and epithelioid sarcoma (57). ‘Undifferentiated/unclassified sarcomas’ is a new category that includes tumors in which no clear line of differentiation could be identified (167). Depending on the predominant cellular morphology, they could be classified as undifferentiated spindle cell, pleomorphic, round cell, or epithelioid sarcoma. Majority of the tumors previously classified as ‘malignant fibrous histiocytoma’ would now be included in this category (567).
The most common non-GIST STS include undifferentiated (pleomorphic) sarcoma, liposarcoma, and leiomyosarcoma (2417). Undifferentiated pleomorphic sarcoma presents as non-specific heterogeneous soft tissue masses with no reported characteristic features on imaging (1), and is not further discussed. We will review liposarcoma and leiomyosarcoma in detail and use them as prototypes to discuss the impact of the new classification and the role of the radiologist in management of sarcomas.
Liposarcoma
The major liposarcoma subtypes are well-differentiated, dedifferentiated, myxoid, and pleomorphic (5). Each of these subtypes is very different from each other on histopathology and has an associated specific radiologic correlate, making the role of the radiologist very useful in diagnosis and follow-up (181920).
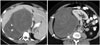
Well-differentiated liposarcoma or atypical lipomatous tumor (ATL) is the most common liposarcoma, and is intermediate-locally aggressive, with a predilection for extremities, retroperitoneum, inguinal, and paratesticular regions (119). The term ATL is used in the extremities where they can be potentially completely resected and essentially cured. However, the term ‘well-differentiated liposarcoma’ is preferred in areas like the retroperitoneum where margin-negative resection is usually not possible (Fig. 2) (5719). It is often difficult to differentiate well-differentiated liposarcomas/ATL from lipomas on imaging, with imaging being sensitive but not specific for the same. Thick/nodular septations and nodular non-lipomatous component in an otherwise fatty lesion is suggestive of ATL (21222324). Large size (> 10 cm) has also been reported to be more common with an ATL (2224). On pathology, in addition to the histological findings, the presence of MDM2 and CDK4 positivity due to 12q13-15 amplification is a useful marker, a finding also seen in dedifferentiated liposarcoma (7).
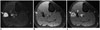
Dedifferentiated liposarcomas are usually deep-seated, with the retroperitoneum being the most common site (likely as these present later) (18). The dedifferentiated component usually resembles pleomorphic sarcoma on pathology (in 90% of cases), and appears on imaging as a nodular non-lipomatous component adjacent to a predominantly fat attenuation tumor (Fig. 3) (181921). These tumors present as (usually) large predominantly fatty masses with a > 1 cm non-lipomatous component(s) that may be soft tissue, fluid or mixed density on CT, the former being most common. The pathological correlate of these different densities is uncertain, but may be due to inflammatory or myxoid component (21). Recurrence can be local in the form of a non-lipomatous lesion with or without fatty component, or as peritoneal sarcomatosis or distant metastases to the liver or lungs (19). A retrospective study of 60 patients observed fluid density lesions to grow faster and often convert to soft tissue density, although this was not statistically significant (21). It is important for the radiologist to pay attention to these non-lipomatous components rather than measuring the entire tumor when following up these patients for restaging or surveillance. Similarly, fluid density should not be ignored as post-surgical in etiology, and should be monitored closely on the follow-up studies as it may represent recurrent disease (19).
Myxoid liposarcoma is the second most common subtype, and unlike dedifferentiated liposarcoma, occurs more commonly in the extremities (181925). It is a cytogenetically simple tumor, demonstrating the t(12; 16) translocation to form the TLS-CHOP oncogene (167). Round cell liposarcoma, previously a separate category, is now understood to represent high grade myxoid liposarcoma and demonstrates the same translocation. The 2013 WHO classification accordingly discarded the term ‘round cell liposarcoma’ from the classification. These tumors are now considered to represent high-grade myxoid liposarcoma, with the prognosis worsening with increasing round cell component (> 5%) (192526).
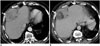
The myxoid component of the tumor appears as fluid density on CT and extremely T2 hyperintense and T1 hypointense on MRI resembling fluid, but unlike cysts, demonstrates contrast enhancement (Fig. 4) (181925). Macroscopic fat is the clincher if observed on imaging, but may constitute < 10% of tumor volume and may be difficult to visualize on imaging (192527). Visualization of a non-fatty non-myxoid enhancing component may correspond to the round cell component, if present, and should be targeted for biopsy (2526). Given the presence of these various components, visualization of a true ‘cyst mimic’ (a tumor which could be misdiagnosed as a cyst without administering intravenous contrast) is uncommon (20). A retrospective study of the MRI features of extremity liposarcomas found lack of encapsulation and presence of solid nodular enhancement to be associated with higher tumor grade in myxoid liposarcomas (20). Presence of peritumoral edema and heterogeneous T2 appearance of the tumor was also found to indicate a more aggressive myxoid tumor and was predictive of the development of pulmonary metastases (20). The metastatic pattern is also unusual in these tumors, with retroperitoneal, opposite extremity, paraspinal, and bony involvement often preceding pulmonary involvement (1927). Bony metastases may be difficult to discern on CT and positron emission tomography (have variable fluorodeoxy glucose uptake), but are T2/short tau inversion recovery (STIR) bright and easily detected on MRI, and whole body MRI may be used for surveillance imaging (2528).
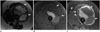
Pleomorphic liposarcoma is the least common and most aggressive subtype of liposarcoma, It most commonly occurs in the extremities, and has poor prognosis if metastatic (181927). It presents as an indeterminate heterogeneous soft tissue mass with absent or minimal fat within it (Fig. 5) (181927). The MRI features often overlap with high-grade myxoid liposarcoma although lack of septations and presence of T2 heterogeneity were found to differentiate pleomorphic liposarcoma from high-grade myxoid liposarcoma (20).
Surgical resection is the treatment of choice for these tumors. Additional adjuvant or neoadjuvant radiation may be given in dedifferentiated, myxoid and pleomorphic liposarcomas (18192129). Ifosfamide and anthracycline-based conventional chemotherapy is used in the neoadjuvant or adjuvant setting, as well as in patients with inoperable or metastatic disease. Trabectedin, a TLS-CHOP inhibitor, was Food and Drug Administration (FDA) approved as targeted therapy for liposarcoma and leiomyosarcoma in 2015, and is effective in myxoid and dedifferentiated liposarcoma (25313233). The role of the radiologist includes accurate tumor diagnosis and mapping for resection, tumor surveillance for recurrence, and response assessment. Apart from a decrease in size, chemotherapy or radiation may also induce the development of macroscopic fat in regions of previous tumor soft tissue (adipocytic maturation), which can be considered as a radiological marker of response (Fig. 6) (303435). The etiology of adipocytic maturation is uncertain, and may be due to treatment induced differentiation in tumor cells, or replacement of tumor cells with mature adipocytes. The clinical significance of this observation is however uncertain, with no definite change in patient prognosis (30343536). Unlike other liposarcomas, pleomorphic liposarcomas often respond to radiation therapy with an increase in size secondary to intratumoral hemorrhage and necrosis, and it is important for the radiologist to be aware of this fact (30). Given the MDM2 and CDK4 positivity of well-differentiated and dedifferentiated liposarcomas, early studies are currently underway to test MTTs against these receptors, with promising initial results (3738).
Leiomyosarcoma
Leiomyosarcoma, the fourth most common sarcoma, can potentially arise from any structure containing smooth muscle, but most commonly arise in the uterus, retroperitoneum, extremities and large blood vessels (394041). Uterine leiomyosarcoma arise de novo from the uterus, with malignant transformation from a pre-existing fibroid being rare (424344). They affect middle to old-aged ladies (> 40 years) and present as large heterogeneous uterine masses on imaging with hemorrhage and necrosis (4245). Differentiating them from degenerating or cellular fibroids is particularly difficult in premenopausal women, as both can show a large size, rapid growth, and a heterogeneous appearance (although these features should be viewed with suspicion in postmenopausal women) (424344). Studies have suggested that MRI features such as ill-defined margins, greater than 50% T2 hyperintense signal, T1 hyperintense foci (due to hemorrhage or coagulative necrosis), low apparent diffusion coefficient values, and early enhancement on the dynamic contrast-enhanced study should be considered suspicious for leiomyosarcomas (45464748). Unlike fibroids, calcifications are consistently absent in leiomyosarcomas (49).
Smooth muscle tumor of uncertain malignant potential (STUMP) is a rare borderline entity that demonstrated non-benign features such as mild atypia or borderline mitotic activity, without any overt features to suggest leiomyosarcoma on pathology (4344). These most commonly arise from the uterus and cannot be definitively differentiated from fibroids or leiomyosarcoma on imaging, and may appear homogenous and T2 dark on MRI or may be heterogeneously enhancing (434445). Tumors may recur as STUMP or leiomyosarcoma (434450). This makes surveillance imaging extremely important.
Retroperitoneal and extremity leiomyosarcomas present as large heterogeneous masses, often with areas of necrosis, cystic change and hemorrhage, although subcutaneous/superficial tumors may appear smaller and homogeneous due to early detection (Figs. 7, 8) (394151). Large vessel leiomyosarcomas most commonly arise from the inferior vena cava (IVC), and may be intra or extraluminal (Fig. 9). The differential diagnosis of a soft tissue lesion arising from a large vessel would be an angiosarcoma or a tumor arising from adjacent structures and secondarily involving the vessel (3952). A recent study described an indiscernible IVC at the point of maximum contact with a retroperitoneal mass to be the most useful feature to predict an IVC leiomyosarcoma, with a negative embedded organ sign useful to exclude the same (53).
Leiomyosarcomas commonly metastasize to the lung, bones, and liver, with intra-abdominal tumors also often involving the peritoneum, and surveillance scans should cover the chest, abdomen and pelvis (394154). Treatment options include conventional chemotherapy, trabectedin and pazopanib (3233). The mechanism of action of trabectedin, which has been approved for use against STS in Europe, is not known but is believed to involve multiple DNA cellular pathways (including secondarily inhibiting the TLS-CHOP oncogene), causing changes in the tumor microenvironment (455). Toxicities include cytopenias, hepatitis, and fluid retention due to capillary leak syndrome leading to anasarca or pulmonary edema on imaging (455). Pazopanib is a VEGF inhibitor and secured FDA approval in 2012 for the treatment of advanced non-GIST non-adipocytic soft tissue sarcomas. Class-specific and drug-specific toxicities associated with it include diarrhea (secondary to colitis), nausea and vomiting, cardiovascular toxicities including hypertension, myocardial dysfunction, and thromboembolism, and hepatitis (5657).
Other Soft Tissue Sarcomas and the Potential Role of MTTs
Given the similar molecular pathways which lead to both carcinogenesis and sarcomagenesis, MTTs which were originally developed for treating carcinomas and common sarcomas like GIST are now being employed for treating rare sarcomas which were considered previously untreatable. In this section, we will discuss a few such sarcomas with specific molecular mechanisms and treatment options.
Imatinib, a small molecule tyrosine kinase inhibitor approved for management of GIST, can inhibit several receptor tyrosine kinases (C-kit, PDGF, colony stimulating factor receptor) and therefore can be effective in other sarcomas which have activating mutations of these kinases. Aggressive fibromatosis (desmoid tumor) is an intermediate-locally aggressive tumor that has a C-kit and platelet-derived growth factor receptor (PDGFR) driven mechanism of sarcomagenesis, and imatinib has been used in its treatment (5859). Imatinib has been found to be effective in dermatofibrosarcoma protuberans, a rare infiltrative cutaneous tumor (256061) which is driven by the t(17; 22) translocation and its resultant COL1A1-PDGFB fusion gene, which activates the PDGFR β receptor (6263). Few early studies have shown the efficacy of imatinib in pigmented villonodular synovitis (56465) which exhibits the translocation t(1; 2) and overstimulation of the macrophage colony stimulating factor gene (666768).
Inhibitors of mTOR, a protein kinase that is integral to the phosphatidylinositol 3-kinase pathway, are FDA approved for second line treatment of metastatic renal cell carcinoma (6970). mTOR also plays an important role in the pathogenesis of some sarcomas notably malignant perivascular epithelioid cell tumors which are uncommon tumors of the retroperitoneum and genitourinary tract (717273). For radiologists, knowledge of the toxicity profile of mTOR inhibitors especially pneumonitis is important as it may result in discontinuation of drug or dose reduction (Fig. 10).
Anaplastic lymphoma kinase (ALK) inhibitors like crizotinib which are FDA-approved for the treatment of ALK-driven non-small cell lung cancer (74) have been found to be effective in inflammatory myofibroblastic tumor (also known as inflammatory pseudotumor), an intermediate-rarely metastasizing tumor that affects children and young adults (575767778). Papillary renal cell cancer is an example of a c-Met driven carcinoma, and targeted inhibitors have shown promising results in early trials (7980). Alveolar soft part sarcoma, an uncommon tumor of the extremities in adolescent with a high incidence of pulmonary and brain metastases (81) has the t(X; 17) translocation activating the microphthalmia transcription factor (MIF) and the c-Met pathway. Clear cell sarcoma is another example of an MIF-driven tumor (82). Both of these sarcomas can be potentially treated with met inhibitors, with a recent phase II trial demonstrating safety of the drug with modest action (83).
CONCLUSION
Since the success of imatinib in treating GISTs, multiple other STS with known molecular mechanism of action are being treated with MTTs. In this era of precision medicine, in order to contribute meaningfully in their multidisciplinary management, it is important for the radiologist to be aware of the variable imaging features of STS, their management with conventional and targeted chemotherapy, and accurate assessment of tumor response and drug toxicities.







 XML Download
XML Download