

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Classification in medicine is central to establishing diagnosis, prognostication as well for selection of treatment strategies. Traditionally, tumors were classified based on anatomical location, for example lung cancer originating in the lung, and within each organ specific group, subgroups were based on the cell type and histopathological features. For example, the 2004 World Health Organization (WHO) classification divided lung cancer into two main subtypes; small cell lung cancer (SCLC) and non-SCLC (NSCLC), and further sub-classified NSCLC into adenocarcinoma and Squamous cell carcinoma (SCC) (1). This classification was sufficient as SCLC was treated with chemotherapy and/or radiation, and NSCLC was primarily treated with surgery, with chemotherapy and/or radiation reserved for unresectable or metastatic tumors. In the past, all advanced NSCLC were essentially treated with platinum based chemotherapy. The radiologist approach to staging and response assessment of tumors also mirrored this classification system. In patients undergoing conventional chemotherapy, response was seen primarily as change in size so size based criteria such as the WHO and Response Evaluation Criteria in Solid Tumours (RECIST) were developed to communicate results in universal fashion.
However, in recent years due to advances in molecular biology and gene sequencing, numerous genes have been identified across hematological and solid malignancies, that when mutated, promote tumorigenesis. Specific genetic mutations in cancers have the potential to be treated with molecular targeted therapies (MTT) such as epidermal growth factor receptor (EGFR) inhibitor Erlotinib in lung cancer, tyrosine kinase inhibitor (TKI) Imatinib in gastrointestinal stromal tumors and vascular endothelial growth factor (VEGF) inhibitor Sunitinib in renal cell carcinoma, leading to dramatic responses and significantly improved overall survival. This has led to a paradigm shift in the classification and treatment of cancer. The last decade saw a slew of newer cancer treatments focused on MTT obtain Food and Drug Administration (FDA)-approval, and treatment options have now exploded. Sub-classification of tumors based on their molecular characteristics allows for stratification of patients for optimal targeted therapy, the promise of precision medicine. Cancer genomics and personalized cancer treatment is the “new kid on the block” and is here to stay.
Lung cancer is the leading example of how transformative cancer classification and care has become in this era of precision medicine. Prior to 2004, the discovery of EGFR mutations in NSCLC and subsequent specific treatment with TKIs, gefitinib and erlotinib, paved the way for precision medicine in NSCLC (Fig. 1). This was quickly followed by the discovery of other driver genes such as echinoderm microtubule associated protein like 4-anaplastic lymphoma kinase 1 (EML4-ALK1) in 2007 and the ALK inhibitor crizotinib, which was granted accelerated FDA approval for treatment of ALK rearranged lung cancers (Fig. 1). EGFR mutations and ALK rearrangements currently account for 12% of all adenocarcinomas in the United States and EGFR accounts for up to 60% of lung adenocarcinomas in Korea and 55% of lung adenocarcinomas in Taiwan (234). The clinical success of targeted agents such as EGFR inhibitors and ALK inhibitors has fueled the research to identify additional genomic abnormalities and targetable agents. Other driver genes in lung cancer include receptor tyrosine kinase (ROS1), fibroblast growth factor receptor 1 and the landscape continues to evolve (Fig. 2). The WHO lung cancer classification was recently updated in 2015 to reflect these advances and testing for EGFR mutations and ALK rearrangements is currently the standard of care for patients with initial diagnosis of lung adenocarcinoma (5). Similar changes have been made to the classification of other cancers for example renal cell carcinoma, breast cancer and gastric cancer, to better reflect the molecular and genomic characteristics. This in turn allows the matching of the right gene with the right cancer agent and eventually improves the efficacy of therapy and patient outcomes.
Imaging plays a central role in the assessment of response to treatment both in day-to-day practice as well as in the setting of clinical trials. Tumors treated with MTT often show morphological changes rather than change in size. Since conventional size based assessment such as WHO and RECIST may not be accurate in this scenario, alternate tumor response criteria such as Choi criteria, Morphology, Attenuation, Size and Structure (MASS) criteria, and Response Assessment in Neuro-Oncology (RANO) criteria have been developed, to capture these morphological changes. In addition, MTT are associated with class specific and drug specific toxicities, different from those encountered with conventional chemotherapeutic agents. A new class of agents that have emerged are the immune checkpoint inhibitors, which promote the body's intrinsic T-cell immune response to cancer cells, and have shown significant activity across multiple cell lines such as melanoma, lung cancer, renal cell carcinoma, bladder cancer and lymphoma. Tumors treated with immune checkpoint inhibitors respond in a different fashion, often demonstrating early increase in size and new lesions prior to dramatic tumor shrinkage, leading to new immune-related response criteria and are also associated with unique range of immune related adverse events. It is important for the radiologists to be familiar with the new cancer classification based on genomics and the various treatment strategies employed, in order to effectively communicate with the oncologists and participate in the multi-disciplinary care.
A review of the entire gamut of genomic alterations in cancer and the changes in classification of different tumors is beyond the scope of this article. Instead, in this paper, we will focus on lung cancer as a prototype of the new molecular classification system and review in detail the two most common targetable alterations in lung cancer. EGFR mutations and ALK rearrangements, and we will look at EGFR mutations and ALK rearrangements beyond lung cancer in different cell lines comparing the presentation and their targeted therapies.
Overview of Epidermal Growth Factor Receptor Mutations and ALK Rearrangements
Epidermal growth factor receptor is a tyrosine kinase receptor of the ErbB family, which is comprised of four related receptors: ErbB1 (EGFR/HER1), ErbB2 (HER2/neu), ErbB3 (HER3), and ErbB4 (HER4). Each receptor is composed of an extracellular ligand binding domain, a transmembrane domain and an intracellular domain (6). When epidermal growth factor binds to the extracellular ligand, a downstream signaling pathway is triggered that includes the Ras-Raf-MAP-kinase and the PI3K-Akt-mTOR pathway, linked to multiple responses promoting tumorigenesis including cell growth, proliferation, motility and survival (7). EGFR induces cancer by one of three mechanisms: mutational activation, amplification and overexpression of ligands. EGFR is overexpressed in multiple human tumors including lung, breast, colorectal, vulvar and head and neck cancers (8).
Anaplastic lymphoma kinase is a member of the insulin ROS1. Once activated it triggers downstream signaling on multiple pathways including RAS/MAPK, PI3K/AKT, JAK/STAT, and Cdc42/Rac (9).
A small inversion in chromosome 2p causes the formation of a fusion gene of the ALK and echinoderm microtubule-associated protein-like 4 (EML4). EML4-ALK rearrangements act as oncogenes and the EML4-ALK fusion protein has oncogenic potential with transforming activity, which is the most common fusion in NSCLC (10). ALK-rearranged cancers are oncogene-addicted with dependence on the oncogene mutation for sustaining proliferation and growth (10). Aberrant activation of ALK has been found in several cancers including NSCLC, anaplastic large cell lymphoma (ALCL), inflammatory myofibroblastic tumor and neuroblastoma (11).
Comparison of EGFR and ALK in NSCLC
EGFR in NSCLC
In NSCLC, EGFR mutations are more common in never smokers, females and patients of East Asian origin (1213). A recent study looking at CT findings and EGFR mutation status, reported that EGFR-mutated adenocarcinoma had significantly higher frequencies of multiple bilateral lung tumors, convergence of surrounding structures, surrounding ground glass opacity, and notch sign at HRCT compared with the non-EGFR-mutated type (1415). Cavitation and pleural effusions less frequent when compared to the non-mutated subtype (1415).
It is important to understand that not all EGFR mutations are responsive to treatment. The two most common mutations of EGFR in NSCLC are deletions in exon 19 and L8585R point mutation in exon 21 (13). These mutations are associated with response to EGFR-TKI treatment and are considered sensitizing mutations, whereas tumors with other EGFR mutation such as exon 20 insertions are resistant to EGFR-TKI treatment. NSCLCs that contain these sensitizing EGFR mutations have been shown to be ‘addicted' to mutant EGFRs for proliferation and survival (1617). Therefore drugs that bind to the EGFR tyrosine kinase and inhibit EGFR are highly effective in treating NSCLC with mutations. Erlotinib and gefitinib are first line EGFR TKIs that reversibly inhibit EGFR and afatinib is a second line EGFR TKI that irreversibly binds to EGFR. All three are administered orally and approved by the FDA as first line therapy in patients with EGFR mutation. Response to EGFR-TKI therapy is usually dramatic with response rates as high as 70%, and associated with early and significant tumor shrinkage (18). A third generation EGFR inhibitor (BI 1482694) got FDA breakthrough therapy designation in December 2015 for metastatic EGFR T790M mutation positive NSCLC (Fig. 1).
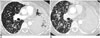
Patients with NSCLC that initially respond to EGFR inhibitors usually have disease progression after a median of 12 months due to the development of resistance (19). In approximately 50% of patients resistance is secondary to a second site mutation T790M at exon 20 (Fig. 3) (20). Afatinib has been shown to be effective against all forms of EGFR including wild type, exon 19, exon 21 L858R and T790M (20), and is currently used as first line treatment in EGFR mutated lung cancer or as second-line agents in patients who develop erlotinib resistance. In 20% of patients EGFR TKI resistance is caused by MET amplification that causing kinase switching triggering the erbB3 pathway instead of the erbB1 pathway (21). MET amplification can coexist with T790M or occur independently, and addition of MET-inhibitors to erlotinib has shown clinical benefit (20).
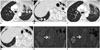
The use of RECIST to determine disease progression in patients on EGFR therapies is under debate. Patients treated with erlotinib may have an initial dramatic response to therapy followed by slow progression over several months suggesting some continued sensitivity to therapy. When erlotinib is discontinued due to disease progression, a flare phenomenon has been observed with dramatic increase in tumor burden, which promptly improves with re-administration of erlotinib (Fig. 4) (22). This ‘flare' phenomenon is not merely an imaging finding but can be associated with symptomatic disease worsening, resulting in hospitalization and in some cases even death (23). The current strategy at many centers is to continue erlotinib beyond RECIST defined progression and add conventional chemotherapy to EGFR inhibitor, or replace erlotinib with second-generation inhibitors such as afatinib.
The most common toxicities that are seen with EGFR inhibitors include skin toxicity, colitis, and rare but potentially fatal pneumonitis. CT findings of pneumonitis include multifocal ground-glass opacities (Fig. 5) with or without interlobular septal thickening or diffuse ground glass changes with consolidation and traction bronchiectasis (2425). Colitis may be manifest on imaging as fluid-filled colon, mural thickening, pericolonic fat stranding, and hyperemic mesenteric vessels. A rare but important toxicity is pneumatosis and pneumoperitoneum, the frequency of which increases with duration of drug treatment (26).
ALK Rearrangements in NSCLC
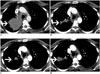
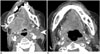
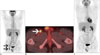
Anaplastic lymphoma kinase rearrangements occur in approximately 2–5% of all NSCLC and are more commonly seen in younger patients (median age early 50s) that are never smokers or former light smokers, adenocarcinoma histology, typically of signet ring cell subtype (27). Metastatic disease at presentation is common, with high propensity for pleuropericardial disease and central nervous system (CNS) metastases (Figs. 6, 7). A multi-institution trial performed for assessing the presence of an ALK molecular phenotype in primary NSCLC on CT found that a central tumor location, absence of a pleural tail and large pleural effusion in patients under 60 years were associated with ALK rearrangements (28). There are multiple ALK rearrangements in NSCLC, of which EML4-ALK is the most predominant (29). Without ALK targeted therapies, these tumors carry a poor prognosis with a median survival of approximately 20 months (30). Crizotinib is a first generation small molecule inhibitor with activity against ALK, MET and ROS1 (31). Crizotinib received accelerated approval from the FDA in 2011 and regular approval in 2013 for ALK rearranged NSCLC.
Tumors treated with crizotinib or other ALK inhibitors often show an initial dramatic tumor shrinkage followed by slow progression on serial studies while the patient remains relatively asymptomatic through progression (32). Acquired resistance to crizotinib occurs secondary to development of mutations in the kinase domain of ALK such as the gatekeeper mutation of L1196M (33). Despite the development of resistance, tumors usually remain ALK-dependent, very similar to EGFR mutant lung cancer on EGFR-TKI therapy, and patients continue to be treated with the crizotinib despite evidence of radiological progression on CT by RECIST.
One of the most common sites of relapse is the CNS, which represents pharmacokinetic failure as crizotinib has poor blood brain barrier penetration (Fig. 6) (34). Surveillance MRI brain is recommended for patients on crizotinib every 6–9 months. Second generation ALK inhibitors developed include ceritinib and alectinib. Ceritinib is an oral small molecule inhibitor of ALK that does not inhibit MET but does inhibit ROS1 and was approved in 2014 by the FDA for patients with metastatic ALK NSCLC that progressed or was intolerant to crizotinib. Alectinib is a highly selective ALK inhibitor that has no activity against MET or ROS1 but does have activity against crizotinib mutations including L1196M and C1156Y and may have a better result in patients with brain metastases (3536).
Side effects that may be seen with crizotinib include development of simple or complex renal cysts, fluid filled bowel loops, peripheral edema, osteopenia and more rarely pneumonitis (Fig. 8) (3738). A peculiar finding is the include development of simple or complex renal cysts, not to be mistaken for cystic metastases or cystic renal cell carcinoma (37).
EGFR Mutations in Non-Lung Cancers
EGFR in Breast Cancer
Between 12–30% of breast cancers have over-expression of ErbB2 (HER2/neu), typically affecting younger patients and have a clinically aggressive course (39). Patients with HER2 expression are more likely to have multifocal cancers, lymph nodal and liver metastases (4041). Tumors that over-express HER2 was previously associated with a poor survival until the development of targeted therapy. Trastuzumab is a humanized monoclonal antibody that works on the extracellular domain of EGFR/HER2, and has shown to significantly improve overall survival in HER2 positive breast cancer (42). Other HER2 targeting agents are pertuzumab, trastuzumab-emtansine (T-DM1) and lapatinib. CNS metastases are more common in patients with HER2 positive breast cancers and subtypes of triple negative patients treated with trastuzumab, likely due to a combination of trastuzumab prolonging survival combined with the inability of trastuzumab to cross the blood brain barrier (42). Trastuzumab may be associated with cardiotoxicity, manifest on imaging as cardiomegaly, pleural effusions and interlobular septal thickening.
EGFR in Head and Neck Cancers
Over-expression of EGFR may be seen in SCCs of the head and neck (SCCHN) (Fig. 9), and correlates with poor prognosis and resistance to radiation therapy (43). Human papilloma virus (HPV) negative SCCHN is associated with EGFR mutations while HPV positive SCCHN is associated with a lack or low expression of EGFR expression (44). Cetuximab is a chimeric human: murine monoclonal antibody that binds to EGFR in the extracellular domain, and thereby prevents the binding and activation of downstream signaling pathways. Cetuximab can also recruit activated immune cells into tumor cells leading to tumor cell death. Cetuximab combined with radiotherapy has been shown to improve median survival in SCCHN (45). One of the common side effects of cetuximab therapy, acneiform skin rash, interestingly correlates with a greater response rate in SCCHN, and may be a surrogate marker of tumor response (45).
EGFR in Colon Cancer
The abnormal expression in EGFR has also been found in metastatic colorectal cancer. There are two anti-EGFR antibodies used in the treatment of colorectal cancer and head and neck squamous cell cancers: cetuximab and panitumab. Cetuximab was discussed previously. Panitumab is a fully human monoclonal antibody (IgG2) also given intravenously. As it does not contain any murine portion of IgG there is less hypersensitivity reactions and it has a longer half-life than cetuximab. Toxicities seen with the anti-EGFR monoclonal antibodies are skin rash, electrolyte abnormalities (hypokalemia, hypomagnesaemia) and an infusion reaction (46). The acneiform skin rash seen with cetuximab as with SCCHN also in colon cancer correlates with a greater response rate in treatment (47). Imaging findings in panitumab toxicities include colitis which may appear as fluid filled colon, mural thickening, pericolonic fat stranding and hyperemia of mesenteric vessels and deranged liver function tests may be associated with periportal edema, gallbladder wall thickening and ascites (25).
EGFR in Vulvar Cancers
Squamous cell carcinoma of the vulva, similar to head and neck cancers can be HPV associated or HPV-independent but the pathogenesis and classification is currently controversial. HPV-associated neoplasms present in younger women with vulvar intraepithelial neoplasm and expression of p16, a cell cycle protein, compared to HPV-independent vulvar SCC occurring in older women with TP53 mutations (484950). A number of mutations have been identified in HPV-independent vulvar SCC including activation of EGFR (51). A study found EGFR amplification in 12% of invasive vulvar SCC and another study found a negative correlation with p16 expression and positive association with p53 raising the possibility of synergism between TP53 and EGFR in the tumorigenesis of HPV-independent vulvar SCC (4851). Studies have also shown EGFR expression in vulvar SCC with advanced stage and lymph node metastases and an association with decrease in survival (5152). While HPV-associated neoplasm in head and neck cancers have shown a better prognosis and therapeutic response, the results are conflicting in vulvar SCC but the presence of EGFR amplification does represent a possible target for therapy with EGFR inhibitors (53).
ALK Rearrangements in Non-Lung Cancer
ALK Rearrangements in Anaplastic Large Cell Lymphoma
Anaplastic large cell lymphoma accounts for 10–15% of pediatric non-Hodgkins lymphoma (NHL) and 1–2% of adult NHL. ALCL more recently has been subdivided into ALK positive and ALK negative subclasses with approximately 90% of pediatric tumors and 40–50% of adult tumors being ALK positive (5455). Most patients present at a late stage with B symptoms with intra-abdominal or mediastinal lymph node involvement and extranodal spread to lung, liver, soft tissue, bone and skin (55). ALK positive ALCL is extremely chemosensitive in front line and relapse with high response rates to traditional NHL chemotherapy regimes (Fig. 10). ALK inhibitors have shown promise in treating relapsed ALK positive ALCL in small phase one trials (56).
ALK Rearrangements in Inflammatory Myofibroblastic Tumors
Inflammatory myofibroblastic tumors (IMT) are mesenchymal neoplasms that occur most commonly in the lung, abdomen and bladder in children and young adults (57). IMT has a wide spectrum ranging from a clinically benign inflammatory pseudotumor to an aggressive sarcomatous neoplasm and surgical resection is the principal treatment. Most cases have a benign clinical course but IMT can recur locally and rarely metastasize (58). Approximately 50% of IMT have ALK rearrangements involving multiple fusion partners (59). ALK expression and rearrangement has been described as a good prognostic marker in IMT with positive cases showing a better outcome compared to a more aggressive course in ALK-negative IMT (5860).
ALK Rearrangements in Neuroblastoma
Neuroblastoma is the most common extracranial tumor in children and is responsible for approximately 12% of pediatric cancer deaths (61). ALK mutations are found in the majority of familial neuroblastoma cases and up to 10% cases of sporadic neuroblastoma (6162). Somatic mutations in ALK have been associated with decrease in overall survival and can occur simultaneously with another genetic aberration that is associated with a poor prognosis (amplification of MYCN) (63).
CONCLUSION
The utility of classification systems ultimately rests on the ability to provide prognostic data and if possible offer specific therapies. In the past, histological classification had little impact on treatment protocol as seen with lung cancer, where almost all patients with advanced NSCLC received platinum-based chemotherapy. In the current era of precision medicine, tumor specimens are tested for targetable mutations and tumors are classified accordingly, in order to match the right patient with the right treatment. As cancer genomics and genomically driven therapies evolve, pure histological classification may become irrelevant in the coming years. As pathologists and oncologists are moving away from the traditional classification systems, as part of multidisciplinary patient management team it is important for radiologist to “adapt” to the evolving changes and “adopt” the new wave of genomic medicine and personalized cancer treatment.




 XML Download
XML Download