

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cystic fibrosis (CF) is a lethal disease induced by mutations in the CF transmembrane conductance regulator (CFTR) gene. CF is one of the most frequently occurring autosomal recessive disorders in Caucasians, with an incidence of 33–50 in 100000 people (1). However, CF is quite rare in the Asian population. The incidence of CF is about 0.3 in 100000 in the Japanese population (2). CF is also rare in Korea, with 10 reported mostly in infants or children (345678910). Although CF has been diagnosed with the standardized sweat chloride analysis, it is now largely diagnosed with the CFTR gene test (178).
Initial radiographic findings of CF include air trapping and linear markings, and subsequent findings include nodular-cystic lesions representing bronchiectasis and mucus plugging (11). On CT, CF manifests as bronchiectasis, cellular bronchiolitis, mucus plugging, and mosaic attenuation (121314). Other diseases can resemble CF clinicoradiologically (1516) and because the disease is rare in Korea, it is difficult to diagnose CF in Korean patients. Therefore, the understanding of clinical and CT features of this rare disease help to stratify patients with clinicoradiologic features suggestive of CF in order to pursue definitive diagnosis with the gene study. The purpose of our study was to describe the clinical and CT features of CF in Korea and compare its features with those of other diseases mimicking CF.
MATERIALS AND METHODS
Our Institutional Review Board approved this retrospective study and waived requirement for patient informed consent.
Study Population
The electronic medical records including CT reports from November 1994 through December 2014 were reviewed to identify patients who underwent a CFTR gene study or met MeSH criteria related to CF. The MeSH words included ‘cystic fibrosis’ or ‘R/O cystic fibrosis’. Twenty-three patients fulfilled these MeSH words, with clinical or radiologic suspicion of CF such as diffuse bronchiectasis especially in children, young adolescents and young adults, recurrent respiratory symptoms or recurrent pneumonia. Of these 23 patients, 10 were excluded because they did not undergo confirmative study with a CFTR gene study or electromicroscopic (EM) biopsy. Finally, 13 patients were included in the study population.
Of these 13 patients (M:F = 3:10; age range, 6–31 years; median age, 16), 10 underwent a CFTR gene study. Of these 10 patients, 7 had CF based on positive gene study. Of the 3 patients who had a negative gene study, 1 was diagnosed with primary ciliary dyskinesia (PCD) by EM biopsy, and 1 was diagnosed with asthma. One patient was not diagnosed with CF, PCD, or asthma, and a final diagnosis of bronchiectasis of unknown cause was made. Three patients without a CFTR gene study underwent EM biopsy and were confirmed to have PCD (Fig. 1).
Currently, 12 CF case reports are published in Korea. Of these 12 CF cases, 5 cases were reported in our institution. Our 7 CF patients were composed of 5 previously reported CF cases and 2 newly reported CF cases.
Clinical Assessment
The medical records were reviewed by one of the authors for the following information: clinical symptoms; family history of respiratory disease; and the results of pulmonary function test (PFT).
CT Acquisition
CT examinations were performed using various multidetector scanners with 16, 40, 64, or 128 channels. Data were reformatted with various section thicknesses for transverse images: 1.25 mm (n = 2), 2.0 mm (n = 5), 2.5 mm (n = 5), and 3.75 mm (n = 1). Scans were obtained from the level of the lung apices to the kidneys. Contrast medium was not injected in any of the patients.
Image Evaluation
Two chest radiologists who were blinded to the final diagnoses retrospectively reviewed CT scans and reached conclusions on all findings in consensus. On CT, parenchymal abnormalities such as the presence of bronchiectasis, cellular bronchiolitis, mucus plugging, consolidation, and mosaic attenuation were assessed. Cellular bronchiolitis was determined based on the presence of centrilobular small nodules or branching centrilobular nodular structures within the secondary pulmonary lobules (17).
Zonal predominance was classified as upper (i.e., above the hilum), lower (i.e., below the hilum), or diffuse in the longitudinal planes. If the diameter of the pulmonary trunk was larger than that of the adjacent ascending aorta or > 29 mm, pulmonary hypertension was recorded. The presence of abnormalities in the upper abdomen were also evaluated and recorded.
RESULT
Demographics and Clinical Characteristics of Patients with CF and Non-CF Diseases
Of a total of 13 patients with suspected CF, 7 (54%; M:F = 1:6, age range; 6–31 years; median age, 15) were confirmed with CF according to the CFTR gene study. Four (31%, M:F = 2:2, age range; 16–28 years; median age, 19) had PCD, a 19-year-old woman had bronchiectasis of unknown cause, and one 8-year-old girl had asthma. Clinical symptoms documented in the 13 patients were as follows: recurrent pneumonia (n = 10), cough (n = 6), sputum (n = 5), and sinusitis (n = 7).
All CF patients underwent PFT. In the CF cases, an obstructive pattern was seen in 3 patients, and a combined pattern was seen in 1 patient. The remaining 3 patients had normal pulmonary function. Only 3 non-CF patients underwent PFT, which showed an obstructive pattern, combined pattern, and normal pulmonary function.
None of the patients with PCD had situs inversus.
CT Findings
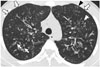
The patterns and distribution of the CT findings were summarized in Table 1. Bronchiectasis, TIB pattern, mucus plugging, and mosaic attenuation were found in all patients with CF. Bronchiectasis and TIB pattern showed a bilateral distribution (Fig. 2). In CF, bronchiectasis and TIB pattern were distributed mainly in the upper lung in 4 (57%) patients (Fig. 3), in the lower lung in 2 patients (29%), and without zonal predilection (diffuse distribution) in 1 patient (14%). In patients with non-CF diseases, bronchiectasis and TIB pattern were distributed with lower lung predominance in 3 patients (50%) (Fig. 4), upper lung predominance in two (33%), and diffuse distribution in one (17%). An area of consolidation was found in 2 of 7 CF patients and in 3 of 6 patients with non-CF diseases, all showed lower lung predominance.
Pulmonary hypertension was suspected in 2 of 7 CF patients and was confirmed by echocardiography. Abnormal upper abdomen findings were found in 2 of 7 CF patients, with one liver cirrhosis and the other fatty replacement of the pancreas. None of non-CF patients showed pulmonary hypertension or upper abdominal abnormalities on CT.
Clinical Follow-Up
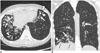
On follow-up evaluation, 4 of 7 CF patients were still suffering from recurrent pneumonia. One patient received lung (double) transplantation and showed no evidence of fatal illness (Fig. 2). One patient died of the disease and fulminant pneumonia. One patient was lost to follow-up.
Literature Review for CF in Korea
In order to study the spectrum of CF in Korean patients, we reviewed the previously identified cases of CF at our institution (n = 5) (3818) and in the literature (n = 7) (4567919), in addition to the cases analyzed in this study (n = 2). Clinical and radiologic features of these patients were summarized in Table 2.
DISCUSSION
Our study showed that CF in Korea manifested as bilateral bronchiectasis, cellular bronchiolitis, mucus plugging, and mosaic attenuation with upper lung predominance, which is consistent with the findings of a previous study (14). The CT and clinical features of CF in our study showed considerable overlap with those of non-CF diseases, especially PCD, with the exception of the zonal predominance.
The predominant CT finding in the early stage of CF is mosaic attenuation due to air trapping related to small airway disease. Other typical CT features include bronchiectasis mainly involving the upper lobes, TIB pattern, and consolidation secondary to mucus plugging (14). In our study, the CT findings of CF showed bronchiectasis, TIB pattern, and mucus plugging, which are consistent with those seen in a Western population. These CT findings also showed upper lung predominance (57%) but less commonly than those seen in a previous study (14).
Non-CF diseases such as PCD may show considerable overlap in both imaging and clinical features (15 leading to difficulty in differentiating CF from PCD based on radiological findings alone. The CT findings of PCD include mucus plugging, bronchiectasis, TIB pattern, air trapping, and consolidation (20). In comparison with CF, the pulmonary lesions in PCD mainly affect the lower lungs (1520). In our study, the CT features of patients with CF and PCD showed substantial overlap, with bronchiectasis, TIB pattern, and air-trapping. The zonal predominance of the findings in CF and non-CF diseases showed no significant difference, inconsistent with the results in Western countries (1420). It is unclear whether this result was due to the small sample size or ethnic differences. Thus, zonal predominance may not be a useful predictor for differentiation between CF and non-CF diseases in Korea.
Most clinical manifestations of CF in our study also overlapped with those of PCD. The most common symptom in CF patients was recurrent pneumonia (86%), followed by cough (57%) and sinusitis (29%). Patients with PCD most commonly presented with sinusitis (100%), followed by recurrent respiratory tract infection (75%). Based on these trends, with a patient's only symptom of recurrent pneumonia, differentiating CF from PCD may be difficult. However, if a patient mainly presents with sinusitis, the suspicion for PCD is greater.
Cystic fibrosis occurs with equal frequency in men and women. The genetic mutation for CF occurs on chromosome 7 and is not impacted by sex. The severity of symptoms related to CF, however, does vary between men and women. Female sex has been associated with greater morbidity and mortality over the course of the disease (21). Our CF population showed a female predominance (F:M = 6:1). A previous study of CF patients aged from 7 to 19 years showed the relative risk of respiratory-related annual hospitalization of 1.38 (95% confidence interval 1.11–1.73) in females as compared to male (22). Female patients show more severe symptoms and a higher rate of hospitalization than male patients, which may be one reason for explaining the female predominance in our CF population. A literature review of CF cases in Korea also showed a female predominance (F:M = 11:3). The median age at diagnosis in our study was older than that reported in Western countries (15 years vs. 6 months) (621). The older age of diagnosis in our study might be explained as follows. First, the suspicion for CF based on clinical findings might be lower because of the low incidence of CF in Korea. Second, there are substantial overlaps in the clinicoradiological features between CF and non-CF diseases. In addition, zonal predominance did not clearly differentiate between CF and non-CF diseases. Based on these considerations, CF should be included in the differential diagnosis of diffuse bronchiectasis, especially in children and young adolescents and female sex with recurrent pneumonia. And thus the CF gene study is recommended for the prompt diagnosis of CF.
Our study had several limitations. First, this was a retrospective study from a single institution, which introduced the possibility of selection bias. In addition, even though we tried to include all cases of potential CF, a number of patients without a definitive diagnosis were excluded. Second, although our study included the largest number of Korean patients, the number of CF patients in this study is still small, as compared with studies from Western countries.
In conclusion, CT findings of CF in Korea showed bilateral bronchiectasis, cellular bronchiolitis, mucus plugging, and mosaic attenuation, which overlaps with those of non-CF patients. The CF gene study is recommended for a definitive diagnosis in patients with these clinicoradiologic features.



 XML Download
XML Download