

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cri-du-Chat syndrome, or "cat-cry syndrome," is due to a large deletion of the terminal short arm of chromosome 5. Therefore, the disorder is also called the 5p-syndrome. Characteristic clinical manifestations of this syndrome are cat-like high-pitched cries, psychomotor retardation, microcephaly and hypertelorism. Cri-du-Chat syndrome is a rare genetic abnormality, and to the best of our knowledge, the associated neuroradiological findings are rarely being reported. We herein report brain abnormalities observed in MRI of a 1-year-old girl with Cri-du-Chat syndrome.
CASE REPORT
A girl aged 1 year and 4 months was admitted for evaluation of psychomotor retardation and an operation for exotropia. She was born at term (gestational age: 40 weeks) by cesarean delivery after an uneventful pregnancy, and was the first child of healthy, unrelated parents. The family history was unremarkable. On admission, she was presented with a characteristic high-pitched cry and microcephaly. She showed delayed motor functions and crawling. She was able to sit unassisted but unable to stand; her developmental characteristics lagged behind for the normal chronologic age. On neurologic examination, she had left leg hypotonia, with normal tendon reflexes and normal cranial nerve signs.
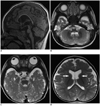
When she was 6 months old, the ventricular septal defect was detected by echocardiography. Chromosome analysis revealed 46,XX,del (5) (p15.2) one month after delivery, resulting in a diagnosis of 5p-syndrome. When the patient was 1-year-old, MRI revealed brain stem hypoplasia (most prominently in the pons), with normal cerebellum, diffuse mild atrophy of the deep white matter of both cerebral hemispheres, as well as mild thinning of the corpus callosum and mega cisterna magna. The anterior limb of the internal capsules on both sides showed reduced myelination, which decreased as compared to normal myelination patterns for 1-year-old infants (Fig. 1).
DISCUSSION
Neuroradiological findings, particularly MRI findings, regarding Cri-du-Chat syndrome are rarely being reported. Currently, there are only six studies reporting MR features of its associated brain abnormalities (1-6). In our case, the MRI revealed hypoplasia of the brain stem, which was most prominent in the pons. Reported neuroradiological findings are mainly brain stem atrophy (with predominant pontine involvement), with or without cerebellar atrophy (1-6). Other associated findings were thinning (or agenesis) of the corpus callosum, mega cisterna magna, fourth ventricle enlargement, lateral ventricle dilatation, cerebellar vermian atrophy (or agenesis), cerebellar cortical thickening and incompleted arborization of white matters (1-6). Our case showed hypoplasia of the brain stem (prominent in the pons) with normal cerebellum, diffused mild atrophy of the deep white matter in both cerebral hemispheres, mild thinning of the corpus callosum and mega cistern magna.
Barkovich et al. (7) suggested developmental classifications of brain stem malformations according to MRI findings. Pontine hypoplasia was the most common finding and was sometimes associated with cerebellar hypoplasia, microcephaly, and corpus callosum anomalies. Cri-du-Chat syndrome was one cause of brain stem hypoplasia. According to previous case reports, including our case, supratentorial abnormalities such as atrophic changes in cerebral white matter and corpus callosum atrophy are often accompanied by pontine hypoplasia and could be explained as secondary changes resulting from brain stem hypoplasia (1-6, 7). Some reports have described skull base malformations and their relationships to brain stem hypoplasia in this syndrome. In a report of skull base malformation in 23 patients with Cri-du-Chat syndrome, sella turcica and clivus malformations and reduced cranial base angles were described (8). The authors suggested that derangements during notochord developments around the specific cranial base region, from ventral neuron migration to the larynx and dorsal development of the rhombencephalic-derived brain stem, pons, and cerebellum, are related to malformations of the cranial base-brain stem and laryngeal region resulting in high-pitched cries in patients with Cri-du-Chat syndrome.
Although brain MRI findings of Cri-du-Chat syndrome are rarely reported, the most common finding was pontine hypoplasia according to previous case reports, as well as our current report (1-6). We suggested that Cri-du-Chat syndrome should be included in a differential diagnosis of brain stem hypoplasia for children with a history of high-pitched cries.
 XML Download
XML Download