

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatic encephalopathy, also known as portosystemic encephalopathy, indicates a spectrum of neuropsychiatric changes in patients with impaired liver function, after an exclusion of other known brain disease. In a patient with chronic liver disease, acute attacks are frequently precipitated by an intercurrent problem, such as gastrointestinal bleeding, acute superimposed hepatitis, concomitant infection, drugs, or constipation, which leads to an elevation of blood ammonia level. Prolonged hyperammonemia results significant brain injury and long-term sequelae, such as intellectual impairment by the primary toxic effects of ammonia on the brain parenchyma. Hepatic encephalopathy is reversible with treatment. Therefore, recognition and correction of hyperammonemia is essential to avoid the complications, as mentioned before (1, 2).
The most common cause of hyperammonemic encephalopathy is acute hepatic dysfunction, and other etiologies include portosystemic shunt surgery, drugs (e.g., sodium valproate, asparaginase therapy, or chemotherapy), infection, hypothyroidism, multiple myeloma, and post-lung or bone marrow transplantation (3).
Although the imaging findings of acute hyperammonemic encephalopathy are well described in children with inborn errors of metabolism (4), the radiologic findings of acute hyperammonemic encephalopathy in adults are less well described. We report a serial MRI of a patient with acute hepatic encephalopathy, who showed cortical laminar necrosis, not reported previously as a consequence of acute hepatic encephalopathy.
CASE REPORT
A 55-year-old man was admitted to the emergency room with generalized tonic-clonic seizures and stuporous mentality, without regaining consciousness for two hours. He was a chronic alcoholic with alcoholic liver cirrhosis. The initial blood ammonia level was 1002 µmol/L (normal range 12-47 µmol/L), and the liver function test results were mildly deranged (bilirubin, 0.91 mg/dL; aspartate aminotransferase, 63 IU/L; alanine aminotransferase, 30 IU/L; pulmonary trunks 1.30 international normalized ratio). Other serum electrolytes and blood glucose levels were within normal ranges (glucose, 113 mg/dL). Anticonvulsant was initially administered intravenously, and the seizures immediately disappeared. Lactulose enema was administered to decrease the patient's blood ammonia level. Electroencephalography performed 3 days after the onset of symptoms showed continuous generalized slowing without epileptiform discharges, suggesting a moderate diffuse of cerebral dysfunction. No hypotensive or hypoxemic episode was observed during the admission days.
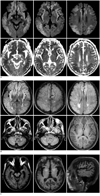
Diffusion-weighted imaging (DWI) (b = 1000), 5 hours after the onset of seizures, showed relatively symmetric bilateral high signals in both temporo-fronto-parietal cortex, including insular and cingulate cortex, with restricted diffusion on the apparent diffusion coefficient (ADC) map (Fig. 1A). The initial differential diagnosis was acute hepatic encephalopathy, hypoxic ischemic change, and other metabolic encephalopathy.
On the fourth day, MRI was performed at 3.0 tesla (Achieva; Philips Medical Systems, Best, the Netherlands), with T2-weighted (TR, 3000 ms; TE, 80 ms), fluid-attenuated inversion recovery images (FLAIR) (TR, 11000 ms; TE, 125 ms; TI, 2800 ms), and T1-weighted inversion recovery (TR, 2000 ms; TE, 10 ms) and intravenous contrast-enhanced images. An interval increase in the extent of brain cortical edema was noted in both cerebral hemispheres, while the occipital cortex and perirolandic regions were uniquely spared. Symmetric high signals were observed in both the medial thalami, basal ganglia, periventricular and cerebellar white matter on the T2-weighted and FLAIR images. Basal ganglia showed subtle high signal on the T1-weighted image in both globus pallidus (Fig. 1B). No pathologic contrast enhancement was seen.
After one month of medical treatment and supportive care, the patient's blood ammonia level decreased to 49 µmol/L, and his seizure attack did not recur. But the patient's intellectual function was impaired. Follow up brain MRI revealed a diffused cortical atrophy in both temporo-fronto-parietal lobe and curvilinear, gyriform high signals on the T1-weighted image in both temporal cortex. In addition, bilateral basal ganglia showed obvious high signals on the T1-weighted image (Fig. 1C). These findings were considered as a cortical laminar necrosis, presumably, the consequence of extreme toxic effect of ammonia.
DISCUSSION
The basic physiology of acute hepatic encephalopathy involves the increased levels of CNS glutamine, as a result of elevated CNS ammonia levels. As CNS ammonia levels increase, glutamine, which is a potent osmolyte, accumulates and leads to numerous consequences, such as astrocyte swelling, oxidative/nitrosative damage, disruption of glucose metabolism, defective neurotransmitter synthesis (such as gamma-aminobutyric acid), and increased blood-brain barrier permeability. As CNS ammonia levels rise with systemic hyperammonemia, the direct toxic effects of ammonia may take hold; thus, increasing CNS ammonia concentrations have been shown to disrupt neuronal function, and glutamine levels have been shown to correlate with the severity of hepatic encephalopathy. These changes collectively contribute to the astrocyte swelling, cytotoxic brain edema, intracranial hypertension and cerebral hypoperfusion, as well as decreased consciousness, which may be seen in an acute hepatic encephalopathy (4, 5). MRI can show cytotoxic brain edema as extensive cortical signal changes, with associated restricted diffusion (3). Similarly, our patient also showed relatively symmetrical, bilateral high signals, in both temporo-fronto-parietal cortex, including the insular and cingulate cortex on the DWI, with restricted diffusion on the ADC map.
Some researches reported the vulnerable or resistant area of the brain in hyperammonemic encephalopathy patients. U-King-Im et al. (3) reported that symmetric involvement of the cingulate gyrus and insular cortex is the striking common imaging finding in adult acute hyperammonemic encephalopathy, while additional cortical involvement is more asymmetric and variable. They suggest that these cortical changes appear to be early imaging finding, and potentially, reversible if aggressive treatment is instituted. They postulate that subcortical white matter, basal ganglia, thalami, and brain stem involvement can also be seen in cases with more severe injury. Several case reports have revealed sparing of the perirolandic and occipital cortex, with early involvement of the cingulate gyrus and the insular cortex in both adults and children patients (4, 6). Thus, the cingulate gyrus and insular cortex appear to be particularly vulnerable to hyperammonemic-hyperglutaminergic encephalopathy, while the perirolandic and occipital cortex seem relatively resistant. However, the pathophysiologic mechanism of regional vulnerability in hyperammonemic encephalopathy is not clearly proved.
McKinney et al. (5) reported abnormalities in the thalami, posterior limb of internal capsule, periventricular white matter, and dorsal brain stem, as well as the diffused cortical involvement on FLAIR and DWI in acute hepatic encephalopathy patients, which can be reversible. The MRI extent on FLAIR and DWI strongly correlates with the maximal plasma ammonia level, and plasma ammonia level correlates well with the clinical outcome. However MRI severity correlates only moderately with the clinical outcome. Our patient showed a periventricular and cerebellar white matter abnormalities, in addition to the diffusede extensive involvement of the cerebral cortex, both basal ganglia, and medial thalami. After 1 month treatment, he showed intellectual impairment, these seem to be caused by extremely high ammonia level on admission. The direct correlation between MRI and clinical outcome has not yet been entirely determined, and it should be explored further in a prospective study.
A follow up brain MRI, in our patient, showed a diffused cortical atrophy in both temporo-fronto-parietal cortex and cortical laminar necrosis in both of the temporal cortex. Cortical laminar necrosis has been described as a consequence of hypoxic encephalopathy, but also in some cases of electrolyte imbalance and diffuse encephalitis. Distinguishing a feature between hypoxic encephalopathy and metabolic encephalopathy is the distribution of the cortical signal changes. Cortical laminar necrosis of hypoxic brain damage is predominant in the watershed zones or the parieto-occipital regions, which possibly reflects the haemodynamic factors (6-8). In our patient, there was no hypotensive or hypoxemic episode during the admission days. Furthermore, the cortical signal change and cortical atrophy were more diffused and generalized in both temporo-fronto-parietal lobes and cortical laminar necrosis were predominant in both temporal cortex. We, therefore, suggest that the cortical laminar necrosis in our case represent an extreme toxic effect of ammonia.
The bilateral basal ganglia of T1 high signal intensity changes have been described in patients with chronic liver disease (1, 9). In our patient, the basal ganglia showed subtle T1 high signal in both globus pallidus on the fourth day after the onset of seizures. This finding can be interpreted as chronic lesions, due to longstanding cirrhosis. On the 37th days after the onset of seizures, a follow up brain MRI showed obvious T1 high signals in the lower portion of both basal ganglia, rather than globus pallidus. It is known that the T1 high signals in the basal ganglia or caudate nucleus can be seen with cortical laminar necrosis in patients of hypoxic-ischemic encephalopathy (10). We suspect the obvious T1 high signals in the lower portion of both basal ganglia by extreme toxic effect of ammonia, as in the cortical laminar necrosis.
In conclusion, extensive cortical edema, including deep gray matter, with sparing of the perirolandic and occipital cortex, appears to be a unique imaging feature, particularly in patients with an extremely high blood ammonia level. In addition, the diffused cortical atrophy with T1 high signals in both basal ganglia and temporal lobe cortex representing cortical laminar necrosis can appear on a follow up brain MRI in such cases. Therefore, we suggest that the brain lesions in our case represent a consequence of toxic effect of ammonia. Acute hepatic or hyperammonemic encephalopathy can be considered as one of the causes in the differential diagnosis of cortical laminar necrosis.
 XML Download
XML Download