

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Lung diseases associated with cystic air spaces include Langerhans cell histiocytosis, lymphangiomyomatosis, bulla, bronchiectasis, and honeycombing. By using paired inspiratory and expiratory high-resolution CT (HRCT) scans obtained at the same anatomic level, changes in lung cysts with respiration can be assessed (1, 2). When the size of cysts changes during respiration, it can be assumed that there is a communication between the cysts and airways. Pathologic specimens show that some cystic spaces are lined by bronchial epithelium or fibrous tissue and some cystic lesions connect with the bronchial or bronchiolar lumen.
To our knowledge, there have been several isolated case reports documenting size changes in cystic air-filled spaces, as seen on expiratory CT (3, 4). Worthy et al. (2) recently reported that the majority of lung cysts become smaller during expiration, but whether their size does in fact change remains a matter of controversy.
The aim of the present study was to determine whether lung cysts change size during respiration in patients with a variety of cystic or destructive lung diseases.
MATERIALS AND METHODS
The study group comprised 54 patients with cystic lung lesions (37 men and 17 women aged 24-76 [mean, 48] years) who underwent paired inspiratory and expiratory HRCT scanning for the assessment of cystic or destructive lung lesions in three university hospitals. The 54 patients had cystic lesions including Langerhans cell histiocytosis (n = 3), pulmonary lymphangiomyomatosis (n = 4), cystic bronchiectasis (n = 13), honeycombing (n = 9), confluent centrilobular emphysema (n = 9), and paraseptal emphysema or bullae (n = 16). Pathologic specimens were obtained in 11 patients: by open lung biopsy in seven patients with Langerhans cell histiocytosis (n = 3) or pulmonary lymphangiomyomatosis (n = 4); by lobectomy in three patients with cystic bronchiectasis (n = 2) or confluent centrilobular emphysema (n = 1); and by necropsy in one patient with honeycombing. Paraseptal emphysema was defined as well-delineated focal peripheral areas of emphysema surrounded by a thin wall and less than 10 mm in diameter. If an area of paraseptal emphysema was greater than 1 cm in diameter, this was defined as a bulla.
Paired inspiratory and expiratory HRCT was performed using a GE Highlight (GE Medical Systems, Milwaukee, WI) or a Somatom Plus-S scanner (Siemens Medical Systems, Erlangen, Germany), with a thin-section (1-1.5-mm collimation) and high-spatial-frequency reconstruction algorithm. End-inspiratory scans were obtained at 10-mm intervals from lung apices to the bases, and end-expiratory scans were obtained at at least five selected levels including the aortic arch, trachial carina, 1 cm below the carina, the inferior pulmonary veins, and 2 cm above the diaphragm. These expiratory CT scans were selected according to the distribution of CT abnormalities. In one case of paraseptal emphysema including bullae, paired inspiratory and expiratory HRCT scans were obtained at the same 10 mm intervals in order to compare the exactly corresponding anatomic level of the cystic lesion. Images were printed with window settings appropriate for viewing the lungs (window width, 1,000-1,500 H; window level, -700 to -600 H).
In the present study, patients were included only if there had been an adequate expiratory effort on each expiratory scan. 'Adequate' was defined as expiration which resulted in a decrease of at least 4 percent in the diameter of the anteroposterior or transverse thoracic cage, as compared with its diameter after inspiration. The diameter of the inner thoracic cage was measured at an anatomically corresponding level on both inspiratory and expiratory scans by two radiologists (K.N.L., S.K.Y.), who reached a consensus.
For each patient, five cystic lesions were selected by the same two radiologists according to the following two criteria: 1) the anatomic level selected was the same for both inspiratory and expiratory scans, and 2) the lesions chosen, in each patient, were the five largest. In this study, a total of 270 cystic lesions were evaluated on paired inspiratory and expiratory HRCT scans to determine whether they decreased in size or remained unchanged on expiration. The longest and shortest diameters were measured using a Vernier caliper on both inspiratory and expiratory scans, and the average thus obtained was taken as representative. Although, in each patient, the five largest cystic lesions were chosen, a lesion was excluded if there was no anatomic correspondence between both CT scans. In such a case, the next largest cyst was selected.
RESULTS
Patients maintained the supine position during full inspiration and expiration, and in all 54, expiration during the acquisition of CT images was adequate. On expiration, the anteroposterior and transverse diameter of the inner thoracic cage decreased by between 4.3% and 15.3%. In patients with confluent centrilobular or paraseptal emphysema including bullae, the decrease in the diameter of the inner thoracic cage was less, varying between 4.3% and 5.3%.
In 52 of 54 patients with cystic lesions (96.3%), either all or the majority of cysts became smaller on expiration (Table 1). Seven (2.6%) of the 270 cystic lesions selected in multiples of five as representative of each diseased lung did not decrease in size. These seven were found in two cases of bullae (2/48; 4.2%) and five of paraseptal emphysema (5/32; 15.6%) (Table 1).
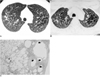
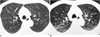
In three patients with Langerhans cell histiocytosis, expiratory scans showed that all cystic lesions became smaller; the mean reduction in diameter was 38% (Fig.1). In four patients with lymphangiomyomatosis, similar scans showed a mean reduction of 45.8% (Fig. 2). In patients with confluent centrilobular emphysema the lesions became smaller on expiration (Fig. 3), while in 73 of 80 cases of paraseptal emphysema (91.3%) including bullae, a size decrease was noted (Fig. 4). In two of sixteen patients with paraseptal emphysema and bullae, no change in size was noted (Fig. 5). In patients with bronchiectasis, the mean reduction in diameter was 39.8% (Fig. 6), while honeycomb cysts became smaller on expiration in all patients (Fig. 7).
DISCUSSION
A variety of lung diseases lead to cystic abnormalities within the lungs. Abnormal air-filled spaces develop in response to diseases which include Langerhans cell histiocytosis, lymphaniomyomatosis, cystic bronchiectasis, honeycombing and confluent centrilobular emphysema, paraseptal emphysema and bullae. Various mechanisms of cystic formation have been proposed, including vascular occlusion or ischemic necrosis, dilatation of the bronchi, disruption of the elastic fibernetwork of the lung, remodeling of the lung architecture, refractile fibrosis, and a check-valve effect in bronchiolar stenosis (5).
When comparing changes in the size of cysts during respiration, it is necessary to consider whether the expiratory effort was adequate, and to this end, intrathoracic anteroposterior or transverse diameters have been measured. In diseases such as emphysema or lymphangiomyomatosis, however, the lung volume does not decrease on expiration, and less change in the diameter of the intrathoracic cage is therefore apparent. In these cases, tracheal or bronchial deformity on respiration may also be considered as respiratory effort. In addition, when comparing the density of normal lung parenchyma surrounding cystic lesions on both inspiratory and expiratory scans, increases in this, while not satisfactorily measuring the adequacy of respiratory effort, may provide some indication of whether it is adequate. In the present study, in order to compare the size of a cyst during respiration, we determined the ratio of the intrathoracic anteroposterior or transverse diameter of the inner thoracic cage during expiratory effort. When comparing inspiratory and expiratory scans, intrathoracic anatomical structures were used as landmarks to confirm that these scans clearly indicated the corresponding anatomic levels.
During expiration, cysts in cases of Langerhans cell histiocytosis became smaller. During the course of this disease, peribronchial granulomatous nodules are replaced by fibrosis, and at a later stage, cysts form. Nodules sometimes have lucent centers, representing the dilated bronchiolar lumen surrounded by granulomas and the thickened interstitium. In some patients, cavitary nodules progress to cystic lesions. As the disease advances, cysts may enlarge to several centimeters in diameter and may have unusual or bizarre shapes. Cysts often appear confluent, joined, or septated, configurations thought to be due to the fusion of several cysts, or because ectatic and thick-walled bronchi are represented (6,7). Cavitary or dilated air-spaces account for cyst change seen grossly, and radiologically, cystic air spaces may communicate with bronchioles.
In four patients with lymphangiomyomatosis, expiratory CT revealed smaller cystic spaces, a finding which differed from that reported in one case of lymphangiomyomatosis associated with tuberous sclerosis. In that case, dynamic thin-section CT demonstrated no change in the size of the cysts (1). Pathologically, lymphangiomyomatosis is characterized by pulmonary interstitial smooth muscle proliferation and cyst formation. Proliferation of atypical smooth muscle occurs mainly around and within the pulmonary lymphatics, venules, and airways. Along the bronchioles, proliferation of spindle cells leads to air trapping and the development of diffuse, cystic dilatation of terminal air-spaces. The changes are progressive and during later stages thin-walled lung cysts are apparent. The mechanism of cyst formation is unknown; a process involving compression of the conducting airways by proliferating smooth muscle in the interstitium has been suggested, though this explanation is controversial (8). Others have postulated that smooth-muscle proliferation within the airways creates a "ball-valve" obstruction, which leads to distention of the terminal air spaces (9). In lymphangiomyomatosis, the decreased size of cysts seen on expiratory HRCT can be explained by their connection with airways.
When progressive centrilobular emphysema becomes severe, at which time it is usually known as confluent centrilobular emphysema, areas of destruction may become larger and confluent. Several respiratory bronchioles may be involved, creating an enlarged space, and it is among the relatively discrete foci that confluence occurs. On HRCT scans the wall of confluent centrilobular emphysema may appear to be thin, probably representing interlobular septa or displaced pulmonary vessels (10). Paraseptal emphysema mainly involves the peripheral portion of the lobule and is therefore most striking adjacent to the pleura, often marginated by interlobular septa. A bulla is defined as a sharply demarcated, air containing space 1 cm or more in diameter with a smooth wall 1 mm or less, and usually occurs in association with emphysema or remote infection. Multiple bullae are related pathogenetically to paraseptal emphysema.
In confluent centrilobular emphysema, expiratory CT shows that cystic lesions become smaller. In several cases, the likelihood that cysts will change their configuration varies with the degree of parenchymal destruction. That is, the more advanced the disease the more likely there will be change in the size of lesions. This may explain why change is observed in cases of confluent emphysema, but not in patchy emphysema. In paraseptal emphysema, including bullae changes in size during the different phases of respiration also occur. A recent report noted that on expiration, the majority of bullae decreased in size (2). In a few cases, however, expiration led to no change in the size of paraseptal emphysema and bullae. Although the mechanism involved in such cases has not been explained, when no change in the size of a cystic lesion is seen on expiration, the abnormality can be considered to be due to bullae (3).
During expiration, honeycomb cysts decrease in size, a change which suggests that these spaces communicate with airways. The size change observed is very similar to that noted in patients with cystic bronchiectasis (3, 4). In patients with interstitial fibrosis, processes such as alveolar disruption, dilatation of alveolar ducts, and bronchiolar dilatation can lead to the formation of honeycomb cysts. The cystic spaces represent dilated respiratory bronchioles (11).
The study has several limitations, one of which relates to the assessment of cyst size: when determining corresponding anatomic levels between inspiratory and expiratory scans, misregistration is possible. To solve this problem, spirometrically gated HRCT or thin-section spiral CT using bone algorithm is required on both inspiratory and expiratory scans, though in practice these modalities are seldom used. Another limitation of our study is that determination of the adequacy of expiratory effort is problematic. This effort depends on the physical condition of the patients involved, and in addition, for each of the conditions mentioned, the severity and extent of pathologic lesions, including cysts, exert a varying influence on the expiratory effort. Although our criterion of expiratory effort was arbitrary, changes in cyst size observed during expiration may reasonably be expected to be equivalent to those which would have been observed if there had been communicated between the cyst and the airways.
We conclude that while in a few cases of paraseptal emphysema and bullae there is no change, the majority of air-filled lung spaces decrease in size on expiration, a finding which suggests that the cysts which became smaller communicate with the airways. To determine whether, for cysts and cystic lesions, this connection does in fact, paired inspiratory and expiratory CT scans are necessary.






 XML Download
XML Download