

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cilostazol is a selective inhibitor of type 3 phosphodiesterase (PDE3) that has been widely used as an antiplatelet agent or a vasodilator [12] through increasing intracellular levels of cyclic adenosine monophosphate (cAMP) and regulating subsequent gene expression mediated by cAMP response element-binding protein (CREB) [3]. Recently, cilostazol has attracted attention because of its reported beneficial effects on the central nervous system, especially its protective actions against neurodegeneration [45]. Phosphorylated CREB (pCREB) promotes neuroprotective gene expression and anti-apoptotic pathways [3]. This suggests that cilostazol may be useful in protecting against excitotoxicity. Therefore, this study investigated the effect of cilostazol treatment on preventing kainic acid (KA)-induced neuronal cell death.
KA, a potent neurotoxic L-analog of glutamate, induces neuronal cell death, primarily in the pyramidal cells of the hippocampus [6]. Animal studies involving KA-induced excitotoxicity have demonstrated that KA induces seizures, glial cell activation, and production of inflammatory mediators [7]. Within hours following a seizure, glial cells are activated, cyclooxygenase (COX) expression is upregulated, and inflammatory processes are initiated [89]. KA systemic administration to rodents resulted in increased activation of glial cells, contributing to the progression of neurodegeneration through the production of inflammatory molecules, such as the iron-siderophore binding protein lipocalin-2 (LCN2) [1011]. LCN2 is secreted by microglia and astrocytes as an autocrine mediator of reactive astrocytosis [12] and is an acute phase protein upregulated in inflammatory brain injuries [13]. Therefore, reducing neuroinflammation and its associated inflammatory mediators are important for the prevention and therapeutic treatment of neuronal cell death. KA-induced neuronal cell death is a useful model for investigating neuroprotective functions of cilostazol.
In the current study, we investigated cilostazol's ability to reduce seizure activity and neuroinflammatory reactions in a KA-induced seizure model. These results suggest that cilostazol may have a therapeutic benefit through reducing glial cell activation.
Go to : 
METHODS
Animals
Male ICR mice, 4 weeks old, were purchased from KOATECH Co. (Pyeongtaek, South Korea) and maintained in the animal facility at Gyeongsang National University (GNU). All animal experiments were approved by the Institutional Board of Research at GNU (GNU-140922-M0046) and were performed in accordance with the National Institutes of Health guidelines for laboratory animal care. Mice were individually housed with an alternating 12 h light/12 h dark cycle and were provided with water and standard chow ad libitum for 1 week prior to experimental procedures.
Drug treatment and seizure induction
Mice were randomly divided to four groups: control group (CTL, n=10), KA-induced seizure group (KA, n=10), 7-day cilostazol pretreatment+KA-induced seizure group (KA+Cilo, n=10), and cilostazol alone group (n=10). Cilostazol (Otsuka Pharmaceutical Co., Ltd, Tokyo, Japan) was diluted with 0.5% carboxymethylcelluose sodium salt (CMC) in normal saline and was administered by intraperitoneal (IP) injection (30 mg/kg) 7 days before KA injection, as previously reported [51415]. Mice were then given an IP injection of KA (30 mg/kg; Abcam, Cambridge, MA, USA) emulsified in 0.9% saline. Mice in the control group received an equivalent volume of 0.5% CMC followed by saline. After KA administration, mice were monitored continuously for 2 h for the onset and extent of seizure activity. Seizure activity was quantitated using a six-point seizure grading scale, as described previously [16]. Animals in all experimental groups were sacrificed at 48 h after KA or control treatment.
Tissue preparation and Cresyl violet staining
For tissue analyses, mice (n=4 per group) were anesthetized with Zoletil (5 mg/kg; Virbac Laboratories, Carros, France) and cardiac perfusion was performed with heparinized saline, followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS). After 6 h of fixation, brains were sequentially immersed in 0.1 M PBS containing 15% sucrose, and then in PBS containing 30% sucrose at 4℃ until the tissues sank to the bottom of the tube. Brains were then frozen in liquid nitrogen and cut into 40 µm thick coronal sections. Sections were stained with Cresyl violet and observed with a BX51 light microscope (Olympus, Tokyo, Japan). Digital images were captured and recorded.
Tissue lysis and western blot analyses
After anesthesia with Zoletil, brains (n=6 per group) were quickly removed from the skull and both hippocampi were dissected and frozen. Tissue lysis for protein extraction was performed as previously described [16]. Protein concentrations were quantified with the Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA, USA) and samples were stored at −80℃. Hippocampal lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by electrophoretic transfer onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). Proteins were immunoblotted with antibodies to glial fibrillary acidic protein (GFAP, 1:3000; Sigma-Aldrich, St. Louis, MO, USA), pCREB (1:1000; Santa Cruz Biotech, Santa Cruz, CA, USA), CREB (1:1000; Santa Cruz Biotech), LCN2 (1:1000; R&D Systems, Minneapolis, MN, USA), COX-2 (1:1000; Santa Cruz Biotech), and transforming growth factor-β1 (TGF-β1, 1:1000; Santa Cruz Biotech). To determine relative protein amounts, β-actin (1:30000; Sigma) was used as an internal control. Specific proteins were visualized using an ECL substrate (Pierce, Rockford, IL, USA). The Multi Gauge v 3.0 image analysis program (Fujifilm, Tokyo, Japan) was used to measure band intensity by densitometry.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay
TUNEL analysis was performed to measure the degree of tissue apoptosis using an in situ cell death detection kit (Roche Molecular Biochemicals, Mannheim, Germany), according to the manufacturer's instructions. Frozen free-floating brain sections were visualized with a confocal microscope (BX51-DSU, Olympus) and digital images captured and documented. TUNEL-positive cells were manually counted in the CA3 region (100×100 µm) in three sections (n=4 per group). The cells were counted by observers blinded to the treatment conditions using 20× objectives.
Triple-immunofluorescence analysis
Triple immunostaining was performed as follows. Free-floating brain tissue sections were incubated simultaneously with mouse anti-GFAP (1:500; Santa Cruz Biotechnology), rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1, 1:150; Wako, Osaka, Japan), and goat anti-LCN2 (1:200; R&D Systems). To distinguish between the mouse primary antibody and endogenous mouse immunoglobulins in the mouse brain, we used the Mouse on Mouse (MOM) immunodetection kit (Vector Laboratories, CA, USA). Samples were then washed three times with 0.1 M PBS and incubated with Alexa Fluor 488-, 594-, and 790-conjugated donkey anti-mouse, anti-rabbit, or anti-goat secondary antibody (Invitrogen Life Technologies, Carlsbad, CA, USA) for 1 h at room temperature. Fluorescence was visualized with a BX51-DSU microscope (Olympus) and digital images were captured.
Statistical analyses
Group differences were determined by one-way analyses of variance (ANOVA) followed by post-hoc analysis with a Student-Newman-Keuls test using GraphPad Prism 5.04 (La Jolla, CA, USA). Student's t-tests were used for grade of seizure activity between two groups. Values were expressed as the mean±standard error of the mean (SEM). A p value <0.05 was considered as statistically significant.
Go to :
RESULTS
Cilostazol reduces seizure activity and hippocampal cell death in KA-induced seizure mice
To assess the effect of pretreatment with cilostazol on neuroprotection in mice with KA-induced seizures, we administered cilostazol at a dose of 10 or 30 mg/kg. However, we found that 10 mg/kg of cilostazol did not significantly reduce seizure activity compared to that in KA-treated mice with 30 mg/kg of cilostazol pretreatment (data not shown). To evaluate whether cilostazol (30 mg/kg) pre-treatment has an effect on KA-induced seizures, seizure activity was monitored for 2 h after IP KA injection (Fig. 1A). Cilostazol pretreatment did not reach seizure scores greater than grade 3 in KA+Cilo mice compared to KA mice 40 min after KA treatment (p<0.05). These results suggest that cilostazol pretreatment attenuates KA-induced seizures. We next examined the effect of cilostazol on neuronal cell death in the hippocampus 2 days post-KA injection. Cresyl violet staining showed that cilostazol pretreatment protected neurons from the seizure-induced neuronal cell death observed in KA-treated mice, specifically in the CA3 region (Fig. 1B). We found that massive loss of KA-induced pyramidal cells was appeared in the CA3 region in KA-treated mice but not in KA+Cilo mice. To evaluate if the observed cell death was apoptotic, a TUNEL assay was performed. The number of TUNEL-positive cells (13.6±1.96) in the hippocampal CA3 region was increased in KA-treated mice compared to that (1.09±0.49) in KA+Cilo-treated mice. This increase in apoptosis was inhibited by cilostazol pretreatment (Fig. 1B). These results demonstrate that cilostazol protects against apoptotic neuronal cell death induced by KA.
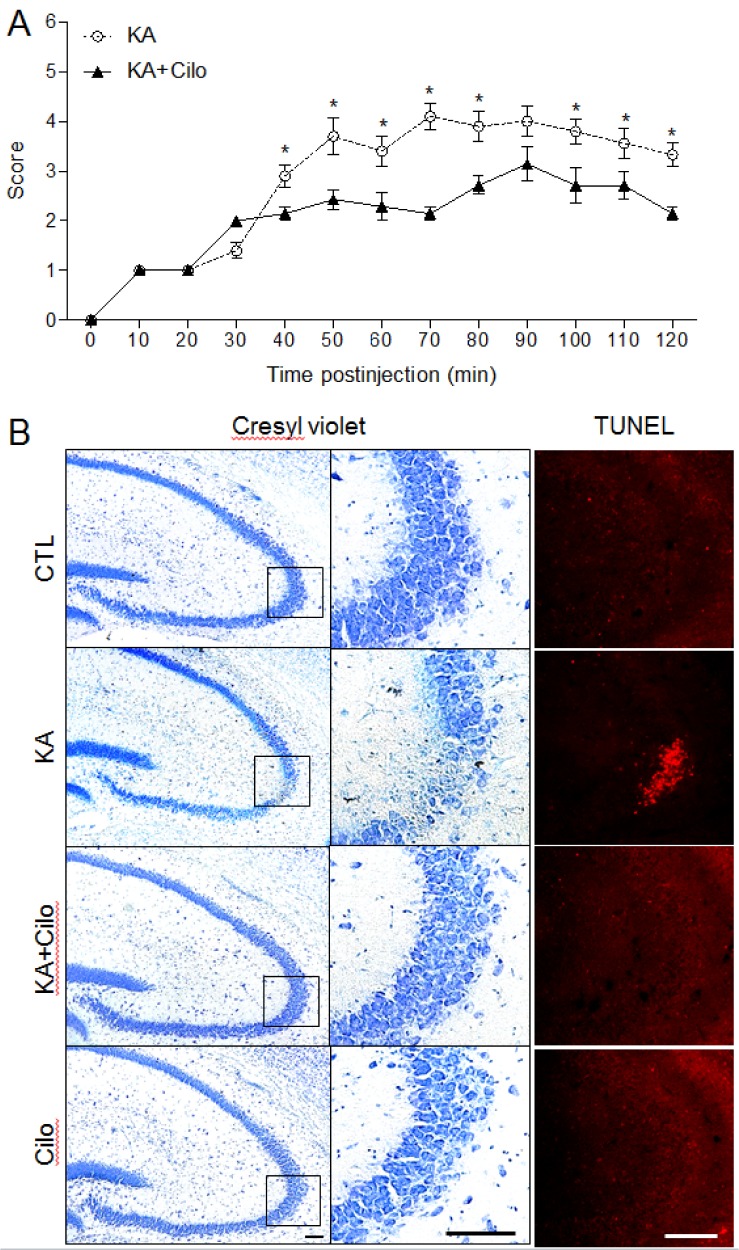 | Fig. 1Effect of cilostazol pretreatment on seizure activity and hippocampal cell death in KA-treated mice.(A) Behavioral seizure scores over time were monitored for 2 h after KA treatment. Data are presented as the mean±SEM. *p<0.05 versus CTL. (B) Representative microphotographs of Cresyl violet and TUNEL staining. Cresyl violet staining shows specific neuronal loss in the CA3 region of KA-treated mice. The areas in black squares in left panels were magnified on the central panels. TUNEL-positive cells indicate neuronal cell death in KA-treated hippocampus. Scale bar=100 µm.
|
Effect of cilostazol pretreatment on pCREB levels in KA-treated hippocampus
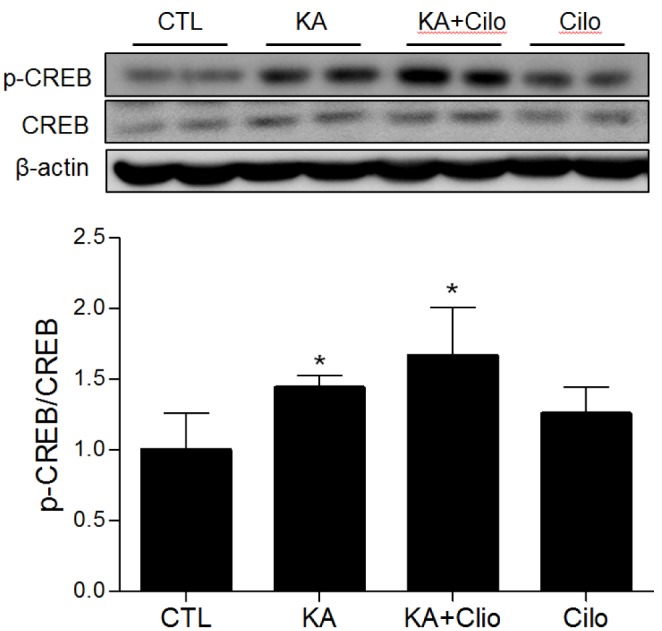
To confirm the association between cilostazol and cAMP-dependent intracellular signaling, CREB signaling activation in the CA3 region of the hippocampus was determined following treatment (Fig. 2). As expected, hippocampal pCREB expression was increased in KA-treated mice compared to controls, as seizure activity is known to increase CREB phosphorylation. In addition, cilostazol pretreatment further induced pCREB expression. Cilostazol activates CREB through preventing degradation of cAMP. Collectively, these results suggest that cilostazol pretreatment may induce neuroprotective effects via enhanced activation of CREB signaling.
Effect of cilostazol pretreatment on hippocampal LCN2, GFAP, and Iba1 expression in KA-treated mice
Glial cell activation induced by an inflammatory response can be identified through detection of immunomodulatory mediators, such as LCN2. To determine if cilostazol pretreatment protected the hippocampus from KA-induced glial inflammation, we examined GFAP and LCN2 expression in activated astrocytes (Fig. 3). Western blots and quantitative protein analysis demonstrated that both GFAP and LCN2 were increased in the hippocampus in KA-treated mice compared to controls (Figs. 3A and B). Immunohistochemistry also revealed intense GFAP and LCN2 staining in the CA3 region in KA-treated mice (Figs. 3C and D). However, these increases were reversed by cilostazol pretreatment. LCN2 staining was particularly seen in the neurons and astrocytes and did colocalize with GFAP-positive astrocytes, but not Iba1-positive microglia (Fig. 3D). In addition, microglial activity was examined through Iba1 expression in the hippocampus. Iba1 immunohistochemical staining results were similar to those for GFAP expression. Our findings suggest that cilostazol protects the hippocampus from KA-induced inflammation by inhibiting glial cell activation.
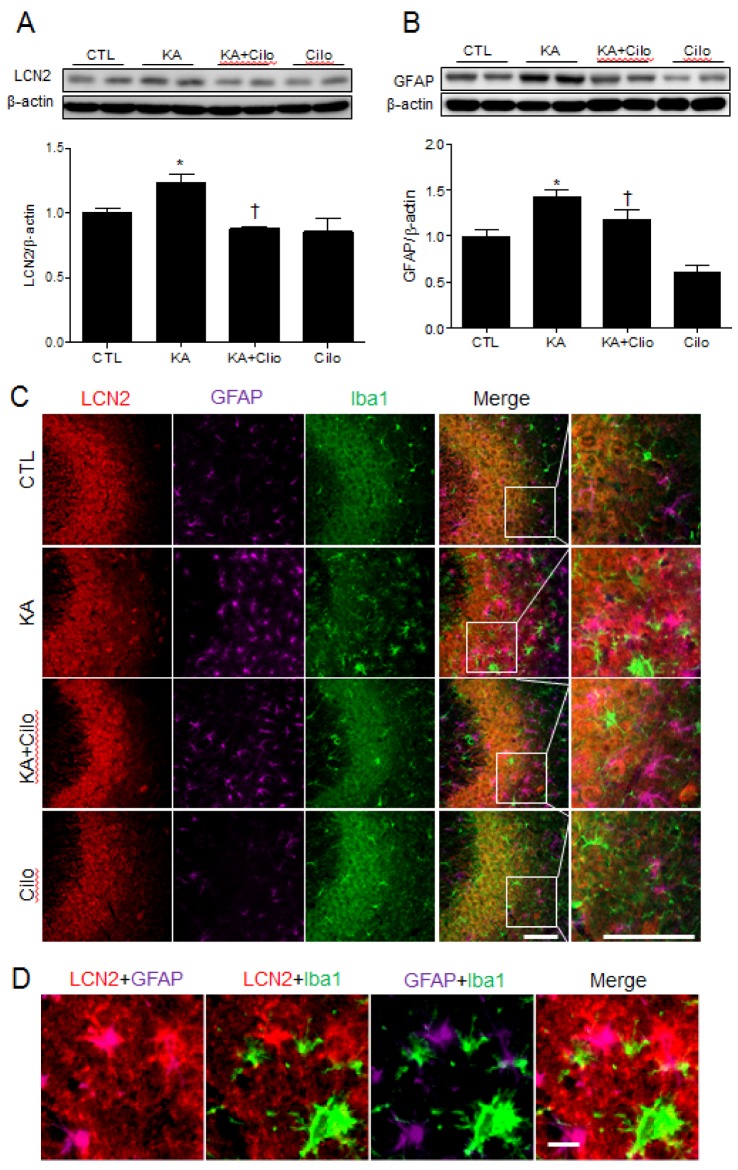 | Fig. 3Effect of cilostazol pretreatment on LCN2 and GFAP expression in KA-treated hippocampus.Western blots and protein quantification of hippocampal LCN2 (A) and GFAP (B). The mean densitometry values were obtained from three separate experiments (n=6 mice per group). Data are presented as the mean±SEM. *p<0.05 vs. CTL. †p<0.05 vs. KA. (C) Representative images of triple-immunofluorescence staining for LCN2 (red), GFAP (purple), and Iba1 (green) in the CA3 region of hippocampus of CTL, KA, KA+Cilo, and Cilo mice. Scale bar=50 µm. (D) Representative images of LCN2 (red), GFAP (purple), and Iba1 (green)-positive cells in the CA3 region of hippocampus of KA-treated mice. Scale bar=10 µm.
|
Effect of cilostazol pretreatment on hippocampal COX-2 and TGF-β1 expression in KA-treated mice
COX-2, a key kinase involved in the inflammatory response, plays an important role in neuronal cell death [17]. TGF-β1 has also been shown to increase in the hippocampus of mice with KA-induced seizures [18]. To assess cilostazol's anti-inflammatory effect in KA-treated hippocampal tissue, western blot analysis was performed to determine COX-2 and TGF-β1 expression (Fig. 4). Cilostazol pretreatment attenuated the KA-induced increase in COX-2 protein expression in the hippocampus (Fig. 4A). In accordance with COX-2 expression, densitometric analysis revealed a significant increase in KA-induced TGF-β1 expression and a reduction to control levels following cilostazol pretreatment (Fig. 4B). Therefore, cilostazol pretreatment attenuated KA-induced neuroinflammatory markers.
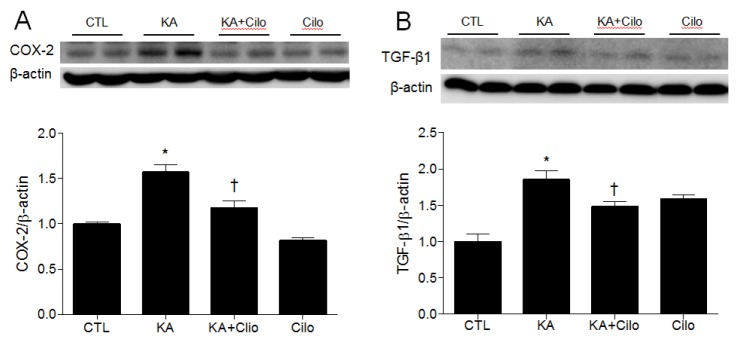 | Fig. 4Effect of cilostazol pretreatment on COX-2 and TGF-β1 expression in KA-treated hippocampus.Western blots and protein quantification of hippocampal COX-2 (A) and TGF-β1 (B). The mean values were obtained from three separate experiments (n=6 mice per group). Data are presented as the mean±SEM. *p<0.05 vs. CTL. †p<0.05 vs. KA.
|
Go to :
DISCUSSION
Several studies have reported that cilostazol acts protectively to activate anti-apoptotic signaling pathways in the brain during neurodegenerative disease [1920]. Here, we evaluated the beneficial effects of cilostazol, a PDE3 inhibitor, on KA-induced excitotoxicity. Cilostazol pretreatment resulted in decreased KA-induced seizure score and reduced apoptotic cell death and glial inflammation in the hippocampus, serving to protect against neuronal excitotoxicity. Both neuronal cell death and glial cell activation were more prominent in hippocampal CA3 regions of KA-treated mice, suggesting that this is the primary lesion site responsible for KA-induced seizures.
Cilostazol's anti-seizure effects were mediated through CREB signaling activation, leading to protection of hippocampal cells. CREB phosphorylation was important for cilostazol's neuroprotection, which has been shown to be induced by PDE3/cAMP and cAMP/CREB signaling through the anti-apoptotic cascade [2122]. CREB is also an important mediator of the biological responses to seizures [23] and seizure activity increases CREB phosphorylation [24]. In the current study, seizure scores and apoptotic neuronal cell death were reduced in the cilostazol pretreatment group, in addition to increased CREB phosphorylation in the hippocampus. CREB activation is known to be regulated by complex phosphorylation and enhancing CREB activity has a neuroprotective role during brain injury [25]. In particular, the present study demonstrated that cilostazol pre-treatment resulted in overexpression of hippocampal pCREB in KA+Cilo-treated mice compared to that in only KA-treated mice. This finding suggests that the neuroprotective role cilostazol might be manifested through subsequent activation of Bcl-2 via pCREB with brain-derived neurotrophic factor (BDNF) expression [1526]. Previous studies also showed that activation of pCREB regulates Bcl-2, which is known to protect against apoptotic cell death after focal ischemia in the rat [27]. Therefore, cilostazol's protective effects on neuronal excitotoxicity could be mediated via CREB signaling, resulting in reduced seizure score.
Glial cell activation, measured by increased astrocyte and microglia activation, has been associated with KA-induced neuronal excitotoxicity [28]. We observed that glial cells become activated after KA injection by upregulation of GFAP and Iba1. Consistent with our result, TGF-β1 is an important cytokine that Morgan et al. [18] showed to be maximal 2 days after KA injection. TGF-β1 is known to regulate astrocyte function. In an astrocytic inflammatory model in vitro, the effects of antiepileptic drug (levetiracetam) showed anti-inflammatory effect of TGF-β1 under pro-inflammatory conditions [29]. Intraventricular injection of TGF-β1 increased GFAP immunoreactivity in the rat hippocampus [30]. Furthermore, TGF-β1 has been protected to protect KA-induced neuronal loss through inhibiting the increase of intracellular Ca2+ [31]. Our findings indicate that anti-excitotoxic effect with cilostazol pre-treatment might stabilize hippocampal TGF-β1 expression against KA-induced excitotoxicity.
Glial cells secretion of LCN2 were increased in our model and LCN2-positive cells were also detected in hippocampal neurons. Increased LCN2 expression could affect iron homeostasis. Neurodegeneration is known to elevate the iron level of neurons [3233]. Consequently, excess iron, catalyzes the formation of free radicals and increases oxidative stress, resulting in neuronal apoptosis [34]. Pretreatment with cilostazol reduced the expression of GFAP, Iba1, and LCN2 in KA-induced seizure mice. These results suggest that cilostazol could ameliorate the inflammatory response through inhibition of LCN2 upregulation. Upon examination of COX-2 expression, we confirmed that cilostazol protects the hippocampus from KA-induced inflammation. COX-2, which facilitates the recurrence of seizures and stimulates hippocampal neuronal loss after KA administration [35], was decreased by cilostazol pretreatment.
Our data provides strong evidence that cilostazol pre-treatment leads to inhibition of neuroinflammation indicated by decreased LCN2 and COX-2 expression. Increased activation of CREB was also observed by cilostazol pretreatment in the KA-induced seizure mice. In addition, the seizure scores and TUNEL-positive cells decreased with treatment, indicating prevention of neuronal apoptosis. Collectively, our results suggest that cilostazol's beneficial effects during seizure may be results of the activation of CREB signaling and reduction of neuroinflammatory reactions. Further studies are needed to dissect the mechanism and therapeutic potency of cilostazol. Other drugs which activate cAMP/CREB, may also be good candidates for the treatment of seizures.
In conclusion, this study demonstrates the neuroprotective effect of cilostazol on neuronal excitotoxicity caused by KA-induced seizures. The results suggest that cilostazol has neuroprotective effects based on multi-target mechanisms through preactivation CREB phosphorylation with anti-apoptotic and anti-inflammatory pathways against KA-induced seizures. Therefore, pretreatment with cilostazol may offer a new therapeutic approach for preventing neurodegeneration.
Go to :
 XML Download
XML Download