

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ovarian cancer is the most lethal in gynecological cancer worldwide, accounting for 200,000 new cases and 125,000 deaths every year [1]. The high mortality of ovarian cancer is associated with the fact that >70% of patients are diagnosed at an advanced stage due to lack of acceptable screening tools for early detection and absence of specific symptoms at early stages [12]. The current standard therapy for advanced ovarian cancer includes tumor-debulking surgery, followed by taxanes combined with platinum (cisplatin/carboplatin)-based therapy. However, over 80% of patients who initially respond to standard chemotherapy eventually develop recurrence with fully chemoresistant disease [3]. Although cisplatin is one of the most effective chemotherapeutic agents for ovarian cancer, the emergence of chemoresistance is a major challenge to successful therapy. Cisplatin is a small-molecule platinum compound, and its anticancer activity is mediated by the formation of intra- and interstrand cisplatin-DNA adducts that activate apoptotic cell death [4]. The mechanisms of cisplatin resistance are not fully understood, but this resistance can result from multifactorial changes at the molecular and cellular levels, including reduced cisplatin accumulation by active efflux or impaired influx, increased detoxification, increased DNA repair, deregulated expression of transporters, and altered expression and activation of genes involved in apoptosis [567]. Recent studies suggest that anticancer drug resistance, including cisplatin resistance, can be mediated by aberrant DNA methylation. In particular, aberrant promoter hypermethylation and subsequent gene silencing can affect sensitivity to cisplatin by inactivating genes that are critical for response to the drug [89].
To identify epigenetically regulated genes directly associated with ovarian cancer cisplatin resistance, we measured the response of 11 human ovarian cancer cell lines to cisplatin and classified them into three groups based on cytotoxicity: sensitive, intermediate, and resistant. We compared expression and methylation profiles of cisplatin-sensitive and -resistant human ovarian cancer cell lines and identified α-N-acetylgalactosaminidase (NAGA) as one of the key genes for cisplatin drug response. NAGA was significantly down-regulated and hypermethylated at a promoter CpG site in cisplatin-resistant cell lines. NAGA is responsible for deglycosylating group-specific component (Gc), a precursor of Gc protein-derived macrophage activating factor (GcMAF). The deglycosylated form of Gc protein cannot be converted into GcMAF and decreased GcMAF levels can promote immunosuppression [10]. Serum accumulation of NAGA in cancer patients has been reported, and serum NAGA activity is correlated with tumor aggressiveness and clinical progression [111213]. However, the involvement of NAGA in the anticancer drug response has not been described. In the present study, using NAGA gain-of-function and loss-of function studies, we demonstrate that the expression of NAGA is regulated by DNA methylationdependent epigenetic mechanisms, which play an essential role in the response of ovarian cells to cisplatin. Additionally, our results suggest that NAGA may be a potential target for sensitizing advanced ovarian cancers to cisplatin-based chemotherapy.
METHODS
Cell culture
The human ovarian cancer cell lines studied were SK-OV-3, ES-2, PA-1, Caov-3, TOV-21G, TOV-112D, OV-90, OVCAR-3, and MDAH 2774, which were purchased from the American Type Culture Collection (Manassas, VA, USA), as well as A2780 and A2780cis, which were purchased from the European Collection of Cell Cultures (London, UK). All cell lines were initially cultured using the media and supplements recommended by the suppliers. Table S1 summarizes the components of culture media for individual cell lines (Table S1). All 11 cell lines were grown as monolayers and attached cells were fully disaggregated by trypsinization between passages. The cell lines were maintained in a 95% humidified and 5% CO2 atmosphere at 37℃.
Cisplatin cytotoxicity assay
The cisplatin cytotoxicity of the 11 human ovarian cell lines (SK-OV-3, ES-2, PA-1, Caov-3, TOV-21G, TOV-112D, OV-90, OVCAR-3, MDAH 2774, A2780, and A2780cis) was determined using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) assays. Briefly, 2×104 cells were seeded onto 96-well plates and incubated at 37℃ overnight. The medium was exchanged with fresh medium containing different cisplatin concentrations (0–100 µM; Sigma-Aldrich; Merck KGaA). After incubation for 48 h, 20 µl of the 2.5 mg/ml MTT solution was added to each well and the plates were further incubated for 2 h at 37℃. One hundred microliters of dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA) was added to solubilize the MTT formazan product by oscillating for 10 min at 37℃. Absorbance at 540 nm was measured using a microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA). Dose-response curves were plotted as the percentage of the control, which was obtained from the sample with no drug exposure. Half-maximal inhibitory concentration (IC50) was calculated as the concentration of cisplatin that reduces cell growth by 50% under the experimental conditions using a nonlinear regression analysis with GraphPad Prism5 software (GraphPad Software, Inc., La Jolla, CA, USA). The 11 human ovarian cell lines were classified into three groups: sensitive, intermediate, and resistant.
Total RNA isolation and mRNA microarray
Total RNA was extracted from eight human ovarian cancer cell lines (SK-OV-3, PA-1, Caov-3, TOV-21G, TOV-112D, OV-90, and OVCAR-3) using the RNeasy mini kit (Qiagen, Inc., Valencia, CA, USA) and amplified and labeled according to the Affymetrix GeneChip Whole Transcript Sense Target Labeling protocol (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The resulting labeled cDNA was hybridized to Affymetrix Human Gene 1.0 ST Arrays (Thermo Fisher Scientific, Inc.). The scanned raw expression values were background-corrected, normalized, and summarized using the Robust Multiarray Averaging approach in the Bioconductor “affy” package (Bioconductor, http://www.bioconductor.org/). The resulting log2-transformed data were used for further analyses. To identify differentially expressed genes, we applied moderated t-statistics based on an empirical Bayesian approach [14]. Significantly up-regulated and down-regulated genes were defined as those with ≥1.5-fold difference in expression level between cisplatin-resistant and -sensitive groups after correction for multiple testing (Benjamini-Hochberg false-discovery rate [BH FDR]-adjusted p<0.01) [15].
Genomic DNA isolation and CpG methylation microarray
Genomic DNA was extracted from eight human ovarian cell lines (SK-OV-3, PA-1, Caov-3, TOV-21G, TOV-112D, OV-90, and OVCAR-3) using the QIAmp mini kit (Qiagen, Inc.), according to the manufacturer's instructions. For genome-wide screening of DNA methylation, the Illumina HumanMethylation450 BeadChip (Illumina, Inc., San Diego, CA, USA) was used, which targets 450,000 specific CpG sites. DNA methylation values were described by β-values, which were determined by subtracting the background obtained from negative controls on the array and calculating the ratio of the methylated signal intensity to the sum of the methylated and unmethylated signals. β-values range from 0 (completely unmethylated) to 1 (fully methylated) on a continuous scale for each CpG site. To identify differentially methylated CpG sites, we calculated the difference in mean β-value (Δβ; mean β-value in resistant group – mean β-value in sensitive group). If the absolute difference in mean β-values (|Δβ|) was >0.3, the sites were defined as differentially methylated CpG sites.
Reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
One microgram of total RNA was converted to cDNA using Superscript II reverse transcriptase and oligo-(dT)12-18 primers (both from Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. RT-qPCR was performed in a 20-µl reaction mixture containing 1 µl cDNA, 10 µl SYBR Premix EX Taq (Takara Bio, Inc., Otsu, Japan), 0.4 µl 50× Rox reference dye (Takara Bio, Inc.), and 200 nM primers for each gene. The primer sequences were: NAGA (forward), 5'-CCCAAGGGTGAACTACAGTCT-3'; NAGA (reverse), 5'-GCTCCACGAACCAATTCAGGAT-3'; GAPDH (forward), 5'-AATCCCATCACCATCTTCCA-3'; and GAPDH (reverse), 5'-TGGACTCCACGACGTACTCA-3'. The reactions were run on a 7500 Fast Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.) at 95℃ for 30 s, followed by 40 cycles of 95℃ for 3 s and 60℃ for 30 s, and a single dissociation cycle of 95℃ for 15 s, 60℃ for 60 s, and 95℃ for 15 s. All reactions were performed in triplicate, and the specificity of the reaction was determined using melting curve analysis at the dissociation stage. Comparative quantification of each target gene was performed based on the cycle threshold (CT) normalized to GAPDH using the 2−ΔΔCt method.
5-aza-2′-deoxycytidine (5-aza-dc) treatment
To demethylate methylated CpG sites, the eight human ovarian cell lines were treated with 10 µM 5-aza-dc (Sigma-Aldrich; Merck KGaA) for three days at 37℃. Each day, the medium was exchanged with fresh medium supplemented with 10 µM of 5-aza-dc.
Transient transfection
To establish a transient expression system, cisplatin-sensitive TOV-112D and cisplatin-resistant SK-OV-3 cells were transfected with pCMV6-AC-NAGA (OriGene Technologies, Inc., Rockville, MD, USA) or pEGFP-N3 (Clontech, Mountain View, CA, USA) DNA constructs using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Briefly, the cells were plated at a density of 6×105 cells/well in 6-well plates containing antibiotic-free complete growth medium and allowed to grow overnight. Two micrograms of each plasmid DNA and 5 µl of Lipofectamine 2000 were diluted separately in Opti-MEM medium (Gibco; Thermo Fisher Scientific, Inc.) to a total volume of 250 µl. The diluted plasmid DNAs and Lipofectamine 2000 were mixed and incubated at room temperature for 20 min to generate the transfection mixtures. The transfection mixtures were added to each well of the 6-well plates, and incubated at 37℃ for 24 h in a 5% CO2 incubator.
Pre-designed small interfering RNA (siRNA) against NAGA (siNAGA, CAT#ID L-011090-00-0005) and a non-targeting control (siNC, CAT#ID D-001206-13-05) were purchased from Thermo Fisher Scientific, Inc. To deplete endogenous NAGA expression, TOV-112D and SK-OV-3 cells were transfected with 100 nM siNAGA or siNC using DharmaFECT 1 transfection reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Knockdown of NAGA was confirmed by RT-qPCR 24 h post-transfection.
The sensitivity of the transfected cells to cisplatin was determined using an MTT assay, described in the “Cisplatin cytotoxicity assay” section.
Western blot analyses
Proteins (30 µg) were resolved using denaturing 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked in 5% skim milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and subsequently incubated overnight at 4℃ with the following primary antibodies: rabbit anticaspase-3 polyclonal antibody (1:1,000, Cell signalling technology, Danvers, MA, USA and mouse anti-α-tublin monoclonal antibody (1:10,000, Sigma-Aldrich; Merck KGaA). After washing, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase for 1 h at room temperature. Chemiluminescence was detected using West Save Star (AbFrontier, Seoul, Korea) according to the manufacturer's protocol. Bands were visualized using an Image Quant LAS mini 4000 (General Electric life sciences, Chicago, IL, USA) and quantified using Image J software (http://rsb.info.nih.gov/ij/index.html).
Statistical analysis
Data are expressed as mean±standard deviation (SD) of ≥3 independent experiments. Statistical analyses were carried out using GraphPad Prism5 software (GraphPad Software, Inc.). An unpaired t-test was used to perform statistical analysis between cisplatin-sensitive and -resistant cell lines. p<0.05 was considered a statistically significant difference.
RESULTS
Differential cisplatin-induced cytotoxicity in 11 human ovarian cancer cell lines
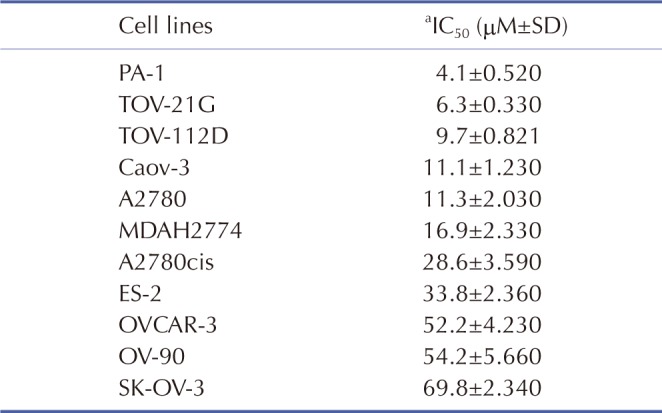
We determined the cytotoxicity of cisplatin in 11 human ovarian cancer cell lines (PA-1, TOV-21G, TOV-112D, Caov-3, A2780, A2780cis, MDAH2774, ES-2, OVCAR-3, OV-90, and SK-OV-3) using an MTT assay. Table 1 illustrates the cytotoxicity of cisplatin in the 11 human ovarian cancer cell lines in order of increasing cytotoxic response: PA-1, TOV-21G, TOV-112D, Caov-3, A2780, MDAH2774, A2780cis, ES-2, OVCAR-3, OV-90, and SK-OV-3. Based on the IC50 values for cisplatin, we classified these cell lines into three groups: sensitive (PA-1, TOV-21G, TOV-112D, Caov-3, and A2780), intermediate (MDAH2774, A2780cis, and ES-2), and resistant (OVCAR-3, OV-90, and SK-OV-3).
Down-regulation of NAGA in cisplatin-resistant cell lines
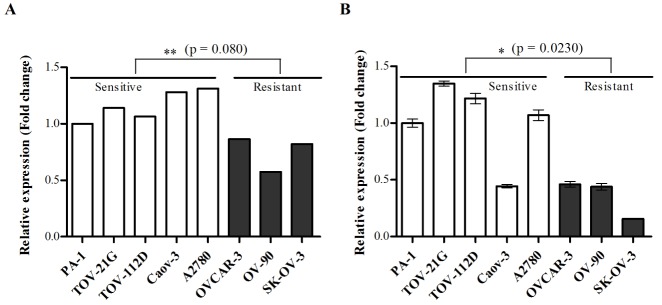
We performed mRNA expression microarray analysis on five cisplatin-sensitive cell lines and three cisplatin-sensitive cell lines. We identified differentially expressed genes by performing moderated t-statistics based on an empirical Bayesian approach [14]. Differentially expressed genes were defined as genes whose levels were up-regulated or down-regulated by at least 1.5-fold between cisplatin-sensitive and -resistant groups after correction for multiple testing (BH FDR-adjusted p<0.01) [15]. NAGA mRNA expression was lower in cisplatin-resistant cell lines compared to levels in cisplatin-sensitive cell lines (Fig. 1A). The downregulation of NAGA mRNA expression was also confirmed by RT-qPCR, which showed significantly lower expression in all cisplatin-resistant cell lines (Fig. 1B).
A CpG site within NAGA promoter region is hypermethylated in cisplatin-resistant cell lines
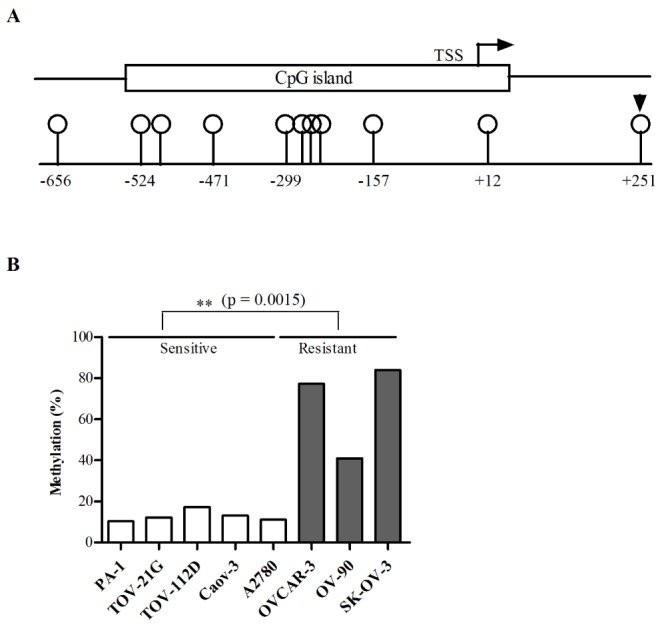
To investigate the mechanism by which NAGA gene expression is regulated in the setting of cisplatin resistance, we performed global DNA methylation profiling of eight human ovarian cancer cell lines using the Illumina HumanMethylation450 BeadChip. To identify differentially methylated CpG sites between cisplatin-sensitive and -resistant cell lines, we applied two significant criteria, the t-test p-values and the difference in mean β-value (Δβ). CpG sites were considered differentially methylated if their t-test p-values were significant at a false-discovery rate threshold of 0.01 [15], and the absolute differences in mean β-values (|Δβ|) were >0.3 at a CpG site within the NAGA promoter region.
The Illumina HumanMethylation450 BeadChip contained 11 CpG sites within the promoter region of the NAGA gene, located between positions 42,071,248 and 42,070,341 of chromosome 22 (human GRCh38/hg38). The 11 CpG sites were at positions −656, −524, −498, −471, −299, −271, −260, −255, −157, +12, and +251 relative to the transcription start site (TSS), as shown in Fig. 2A. Among the 11 promoter CpGs, the CpG site located at +251 from the TSS was significantly hypermethylated in cisplatin-resistant cell lines compared with cisplatin-sensitive cell lines (Fig. 2B).
NAGA gene expression is regulated by epigenetic modification
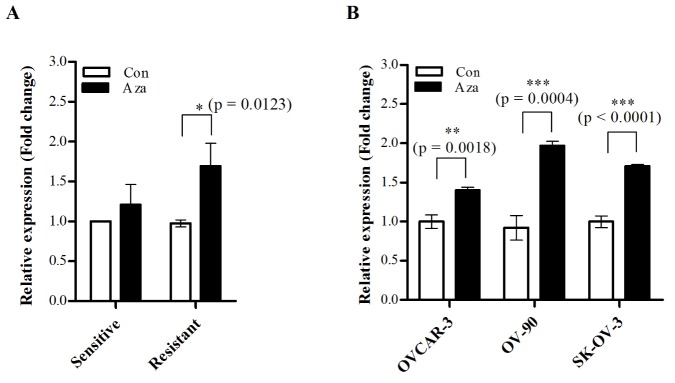
Next, we assessed whether NAGA expression is controlled by epigenetic regulatory mechanisms using a DNA methyltransferase inhibitor. Eight cisplatin-sensitive and -resistant cell lines were treated with 5-aza-2′-deoxycytidine, and NAGA
mRNA expression was quantified by RT-qPCR. After treatment with 5-azadc, significantly increased NAGA expression was detected in the cisplatin-resistant cell lines, but not in the cisplatin-sensitive cell lines (Fig. 3A). NAGA expression level was restored by treatment of 5-aza-dc in all three cisplatin-resistant cell lines, but remained unaffected in untreated cells, indicating that NAGA expression is epigenetically silenced by DNA methylation in cisplatin-resistant cell lines (Fig. 3B).
Modulation of NAGA expression affects cisplatin sensitivity in human ovarian cancer cells
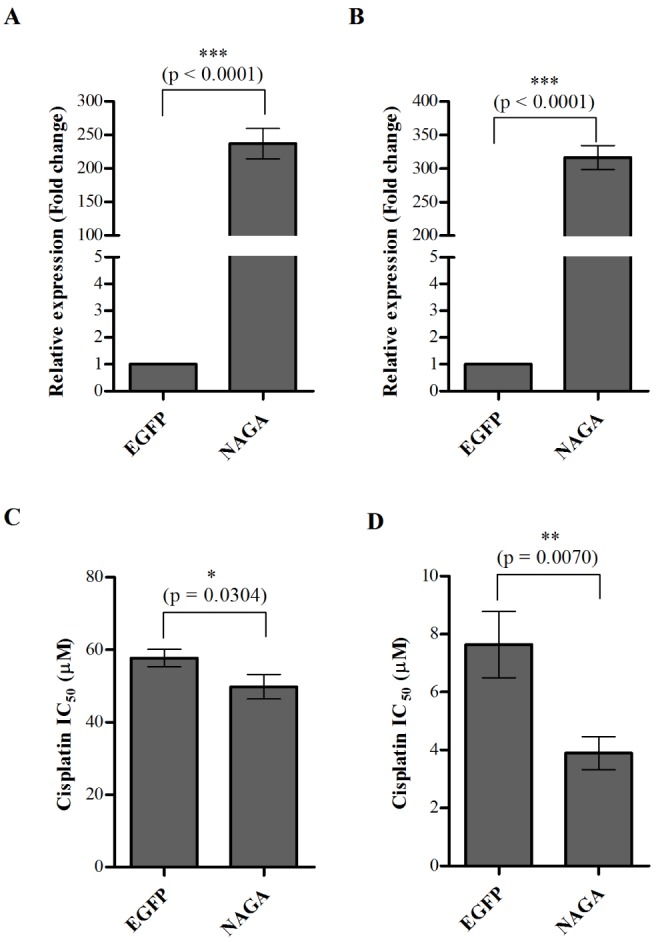
We investigated whether NAGA overexpression could enhance sensitivity of ovarian cancer cells to cisplatin. Cisplatin-resistant SK-OV-3 cells and cisplatin-sensitive TOV-112D cells were transiently transfected with NAGA or EGFP expression plasmids. Following a 24-h transfection, the expression levels of NAGA were analyzed using RT-qPCR. As shown Figs. 4A and B, the expression of NAGA mRNA was increased ~258-fold in NAGA-transfected SK-OV-3 cells and ~317-fold in NAGA-transfected TOV-112D cells, compared with EGFP- mock transfected cells (Figs. 4A and B). The cytotoxicity to cisplatin was also determined in EGFP- or NAGA-transfected cells using an MTT assay. Overexpression of NAGA lead to a significant decrease (~13%) in the IC50 of cisplatin in NAGA-transfected SK-OV-3 cells compared with EGFP-mock transfected cells (Fig. 4C). Similarly, the cisplatin IC50 value dropped by ~49% in NAGA-transfected TOV-112D cells (Fig. 4D). These results indicate that overexpression of NAGA sensitizes human ovarian cancer cells to cisplatin.
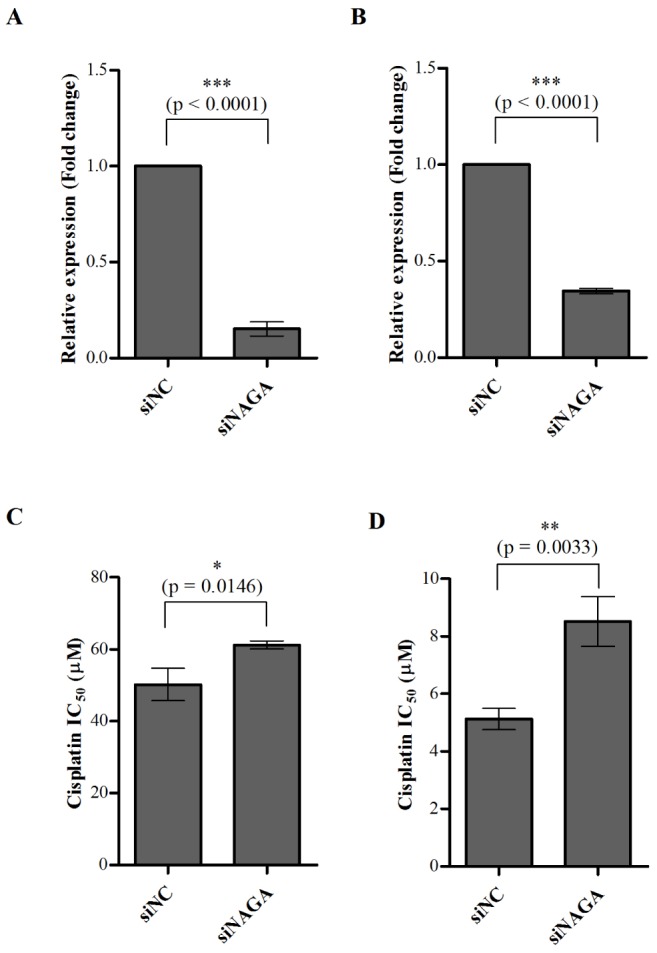
We further explored how NAGA expression affects cisplatin resistance in ovarian cancer cells using siRNA-induced NAGA knockdown. After 24 h transfection, the knockdown of endogenous NAGA expression was confirmed by RT-qPCR. The expression of NAGA mRNA was reduced by ~89% in siNAGA-transfected SK-OV-3 cells and ~67% in siNAGA-transfected TOV-112D cells, compared with siNC- mock transfected cells (Figs. 5A and B). The cisplatin resistance of siRNA-transfected ovarian cancer cells was then evaluated using an MTT assay. Knockdown of NAGA expression induced a significant increase in the cisplatin IC50 value by ~22% and 66% in SK-OV-3 cells and TOV-112D cells, respectively (Figs. 5C and D).
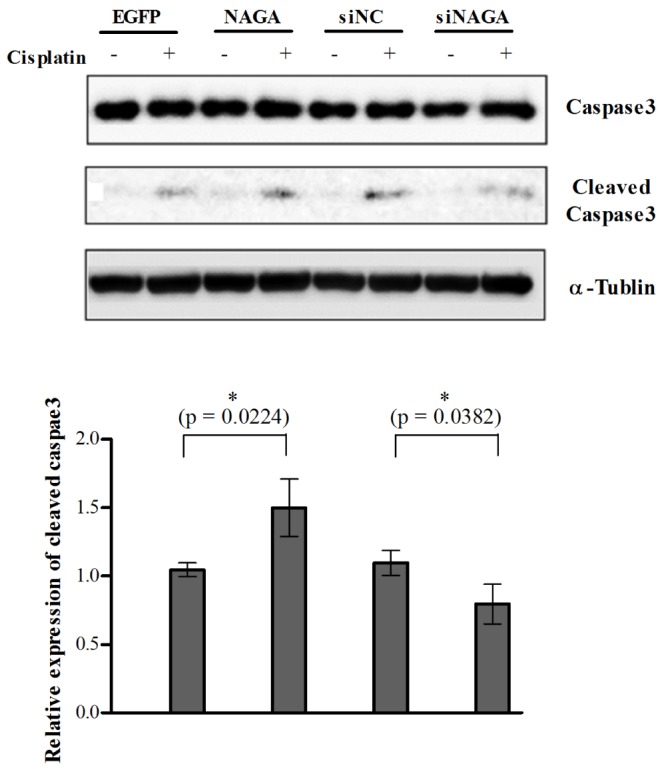
The cleavage status of caspase 3, an indicator of apoptosis was determined in the NAGA-overexpressed SK-OV-3 cells after treatment with cisplatin (30 µM for 10 h). Cisplatin-induced caspase 3 cleavage was increased by the overexpression of NAGA (Fig. 6), correlating with enhanced cytotoxicity observed in MTT assay (Fig. 4C). Decreased caspase 3 cleavage was also detected in the siNAGA-transfected SK-OV-3 cells compared with siNC-mock transfected cells (Fig. 6).
To determine whether the mitochondrial pathway was involved in cisplatin-induced apoptosis in SK-OV-3 cells, we examined changes in mitochondrial membrane potential. As shown in Fig. S1, the mitochondrial membrane potential did not significantly change in SK-OV-3 cells after 8 h of cisplatin treatment regardless of the level of NAGA expression (Fig. S1). These results indicate that cisplatin induces apoptotic cell death via mitochondrial-independent pathway in cisplatin-resistant SK-OV-3 cells.
Taken together, our data suggest that NAGA is involved in the regulation of cisplatin sensitivity by promoting apoptosis in human ovarian cancer cells.
DISCUSSION
Previous studies have been reported that aberrant changes in DNA methylation play an important role in the development of cisplatin resistance in human cancer cells, including ovarian cancer. In particular aberrant hypermethylation of CpG sites within promoter regions can affect the sensitivity of cancer to chemotherapy by inducing silencing of genes responsible for drug response [1617].
By comparing expression and methylation profiling of cisplatin-sensitive and -resistant human ovarian cancer cell lines, we identified a candidate gene, NAGA, as a key regulator of the anticisplatin drug response. NAGA mRNA expression was significantly down-regulated in cisplatin-resistant ovarian cancer cells, which was associated with hypermethylation of a CpG site within the NAGA promoter, located +251 from the TSS. Reduced NAGA expression in cisplatin-resistant cell lines was restored by treatment with the DNA demethylation agent, 5-aza-dc, indicating that NAGA transcriptional silencing occurs through hyper-DNA methylation of the NAGA promoter. Furthermore, restoration of NAGA expression resulted in increased cytotoxicity to cisplatin, whereas NAGA loss caused increased cisplatin chemoresistance in both cisplatin-sensitive and -resistant cell lines. In addition to NAGA, another glycogene GBGT1 encoding the globoside alpha-1,3-N-acetylgalactosaminyltransferase 1 has been reported its epigenetic regulation in ovarian cancer. The GBGT1 expression was silenced by promoter hypermethylation and inverse correlation between DNA methylation and expressions of mRNA and protein was confirmed in ovarian cell lines and primary ovarian cancer tissue samples [18].
Cisplatin exerts its cytotoxic effects mainly through apoptosis and alteration or defiance against apoptotic signaling process could confer cisplatin resistance. There are two major types of cell death pathways, namely, the extrinsic pathway and the intrinsic pathway. The extrinsic pathway is initiated when ligands bind to the death receptors such as tumor necrosis factor-α (TNFα) receptor 1, CD95 (Fas) and TNF-related apoptosis inducing ligand (TRAIL) followed by receptor clustering and recruitment of adaptor molecules into a death-inducing signaling complex (DISC), which then activates an initiator caspase, procaspase 8. Activated caspase 8 then propagates the apoptotic signal via the activation of the executioner caspase 3. The intrinsic pathway also known as mitochondrial pathway is initiated by a variety of receptor-independent stimuli such as DNA damage which trigger mitochondrial membrane permeabilization and subsequently result in the release of proapoptotic mitochondrial proteins and cytochrome c into cytosol. The initiator caspase procaspase 9 is activated through the dimerization with apoptosis promoting activating factor-1 (APAF-1) and formation of an active apoptosome complex containing cytochrome c, APAF-1 and caspase 9. Bcl-2 family proteins regulate DNA damage-induced apoptosis by preventing formation of mitochondrial pores and inhibiting the release of mitochondrial cytochrome c. [1920]. It has been reported that p53 play a critical role in cisplatin-induced cell death via two mechanisms, transcription-dependent and -independent pathway. In the transcription dependent pathway, p53 upregulates the expression of pro-apoptotic proteins such as PUMA, Bax and BID which are involved in the regulation of the intrinsic pathway and activates death receptors such as CD95, DR5 receptors which mediate the extrinsic pathway. In addition, p53 can also suppress anti-apoptotic proteins such as Survivin. In the transcription-independent pathway, p53 localizes to the mitochondria following cytotoxic insults where it interacts with Bcl-2 and Bcl-XL and inhibits their anti-apoptotic function at the outer mitochondrial membrane. In addition to mitochondrial p53, cytosolic p53 induces the activation of Bax, subsequently resulting in mitochondrial membrane permeabilization and cytochrome c release [21].
The caspase 3 assay and mitochondrial permeability detection assay showed overexpression of NAGA promoted cisplatin-induced apoptotic cell death, but the mitochondrial pathway was not involved in cisplatin-induced apoptosis in p53 null SK-OV-3 cells. Together, our results suggest that NAGA plays an essential role in sensitizing ovarian cells to cisplatin by promoting apoptotic cell death.
NAGA is a lysosomal enzyme that is responsible for the deglycosylation of Gc, also known as vitamin D-binding protein, a precursor of GcMAF. Deglycosylated Gc protein cannot be converted into GcMAF, leading to defective macrophage activation [10]. Increased serum NAGA activity has been reported in variety of cancer patients, which is associated with clinical severity [111213].
In the present study, we provide novel evidence that NAGA acts as a cisplatin sensitizer and its gene silencing via hypermethylation of a CpG site within its promoter confers cisplatin resistance in ovarian cancer. Our findings suggest that NAGA is a new potential therapeutic target for the treatment of chemoresistant ovarian cancer.
 XML Download
XML Download