

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Vascular smooth muscle cells (VSMCs) are one of the cell types constituting the arterial wall, and their proliferation plays an important role in the progression of early atherosclerosis and vascular restenosis [1]. Additionally, the viability of VSMCs is known to be an important factor in the rupture of arteriosclerosis [2]. The atherosclerotic plaque rupture occurs myocardial infarction and stroke through thrombus formation [3]. In this process, various cytokines [4] and growth factors [56] induce vascular inflammation and remodeling. Platelet-derived growth factor-BB (PDGF-BB) is a growth factor that is increased in the blood vessels of cardiovascular disease [7]. It is also known to be a powerful stimulation factor of proliferation or migration of VSMCs [8]. Most treatments for cardiovascular diseases, such as vascular restenosis and arteriosclerosis, are performed through surgical operations. The stent is coated with a microtubule-targeted drug such as paclitaxel for the prevention of vascular restenosis [9]. After the surgery, thrombosis and approximately 12% angiographic restenosis occur [10]. Michigan Cancer Foundation-7 (MCF-7), one of the breast cancer cell lines, is known to be resistant to apoptotic cell death by paclitaxel through increased stimulation of the autophagic pathway [11]. Therefore, it is expected that angiographic restenosis after angioplastic surgery with a microtubule target agent-eluting stent may be associated with autophagy. However, insufficient studies have reported the effects of microtubules and autophagy in VSMCs.
Autophagy contributes to the maintenance of intracellular homeostasis through recycling or degradation of intracellular elements [1213]. The targeted proteins of autophagy are surrounded by double-membraned vesicles called autophagosomes [14]. The autophagosome fuses with lysosomes, and then forms autolysosome in which intracellularly damaged organs and proteins are decomposed [15]. In vascular cells, autophagy acts as a regulator of cell viability and function [1617]. It also prevents the abnormal function of blood vessels, such as inflammation, by protecting against potential damage by various stresses [18]. Autophagy in VSMCs is stimulated or inhibited by various stimuli and stress factors (metabolic stresses, reactive species, cytokines, and drugs) [19]. It has been reported that autophagy in VSMCs promotes senescence, neointima formation, and atherogenesis [20]. However, it has also been reported that the promotion of autophagy inhibits neointima formation induced by the balloon injury model [21]. Together, these findings indicate that differences in the autophagy levels of VSMCs may be completely different phenomena (regulation of VSMC homeostasis). In the treatment strategy of cardiovascular disease by VSMC regulation through inhibition or promotion of autophagy, there remains a lack of evidence to determine which direction is the right strategy. The results of the present study suggest that the strategy of increasing autophagy is valid for the treatment of atherosclerosis and restenosis.
METHODS
Materials
Cell culture materials were purchased from Invitrogen (Carlsbad, CA, USA). PDGF-BB was obtained from PeproTech (Rocky Hill, NJ, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), bafilomycin A1, rapamycin, and 3-methyladenine (3-MA) were purchased from Millipore Corporation (Billerica, MA, USA). N-Acetyl-L-cysteine (NAC), 2′,7′-dichlorofluorescin diacetate (H2DCFDA), paclitaxel, and vinorelbine were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Anti-β-actin, anti-microtubule-associated protein light chain 3 (LC3) A/B, anti-α-tubulin, and anti-caspase-3 antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Cell culture
Rat aortic VSMCs were obtained from Cell Applications Inc. (San Diego, CA, USA). Rat aortic VSMCs were cultured in DMEM media containing 10% (v/v) fetal bovine serum (FBS), 100 IU/ml penicillin, 100 µg/ml streptomycin, 4.5 g/L D-glucose, 110 mg/L sodium pyruvate, and 2 mM L-glutamine at 37℃ in a humidified atmosphere of 95% air and 5% CO2. VSMCs were used at passages 4-9. Before all assays, VSMCs were maintained under serum-starved conditions.
Cell proliferation and viability assays
To evaluate the effects of paclitaxel and vinorelbine on cell proliferation, the proliferation and viability levels of PDGF-BB-stimulated VSMCs were determined using the MTT assay [22] and cell counting assay [23], respectively, as described previously. Briefly, for MTT assay, VSMCs were seeded in 96-well plates at 3.5×104 cells/ml and were cultured in DMEM containing 10% FBS at 37℃ and 5% CO2 for 24 h. After incubation in serum-free DMEM for 24 h, the cells were pretreated with 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, 0.2 µM rapamycin, or 5 mM NAC for 24 h, followed by treatment with or without 25 ng/ml PDGF-BB for another 24–72 h. At the end of the reaction, the medium was removed, and 200 µl of MTT solution was added to each well for 4 h. After the MTT solution was replaced with 200 µl of dimethyl sulfoxide (DMSO), the absorbance was measured at 565 nm using a microplate reader (Infinite M200 PRO; Tecan Group Ltd., Zürich, Switzerland). For cell counting assay, VSMCs were seeded into 12-well plates at a density of 1.6×105 cells per well and were incubated for 24 h. After incubation in serum-free DMEM for 24 h, the VSMCs were pretreated with 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, 0.2 µM rapamycin, or 5 mM NAC for 24 h. Next, the VSMCs were further cultured in medium with or without 25 ng/ml PDGF-BB for 48 h, and the VSMCs were counted using a hemocytometer.
Reactive oxygen species (ROS) generation assay
ROS production was determined as described previously [2224]. H2DCFDA was used to evaluate the intracellular ROS level. VSMCs (1×105 cells/ml) were pretreated with or without 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, and 0.2 µM rapamycin for 24 h, followed by the addition of 25 ng/ml PDGF-BB for 48 h. The cells were stained for 30 min at 37℃ by 20 µM H2D-CFDA. The intracellular ROS level was determined by measuring the fluorescence intensity using a microplate reader (Infinite F200 PRO, Tecan Group Ltd., Männedorf, Switzerland).
Western blot analysis
Western blotting was performed as described previously [22]. Total cell lysates were prepared using RIPA lysis buffer (150 mM sodium chloride, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 50 mM Tris-HCl, pH 7.5, and 2 mM EDTA) containing proteinase inhibitors. After 10 min incubation on ice, cell lysates were centrifuged at 13,000 rpm for 10 min, and the supernatants were collected. The protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA). The proteins were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to a polyvinylidene fluoride membrane (ATTO Corporation, Tokyo, Japan). After blocking, the membranes were incubated with primary antibodies to determine the levels of LC3A/B (1:1000) and caspase-3 (1:1000). The membrane was washed and incubated with a corresponding horseradish peroxidaseconjugated secondary antibody, and the signals were detected using an ECL western blotting detection system. The activated proteins were normalized against β-actin levels. Band intensities were quantified using Quantity One software (Bio-Rad, Hercules, CA, USA).
Immunofluorescence staining analysis
Immunofluorescence staining was performed as described previously [22]. VSMCs were seeded on cover slips and were incubated for 24 h, followed by starvation in serum-free medium for 24 h. The serum-starved cells were pretreated with 1 µM paclitaxel or 0.2 µM vinorelbine for another 24 h. Following stimulation, the cells were treated with 25 ng/ml PDGF-BB for 48 h, and then were washed twice in cold phosphate-buffered saline (PBS). The cells were fixed with cold 4% formaldehyde for 10 min and were permeabilized with chilled 0.25% Triton X-100 for 2 min. Next, a blocking step was performed using 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature. The cells were incubated with primary anti-α-tubulin antibody for 1 h, followed by incubation with FITC-labeled secondary antibody for 1 h. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and immunofluorescence images were obtained using a confocal laser scanning microscope (LSM5 LIVE; Zeiss, Oberkochen, Germany).
Statistical analysis
All data are expressed as means±standard error of the mean (SEM) of three independent experiments. Analysis of variance (ANOVA) was used to compare parameters among multiple groups (GraphPad, San Diego, CA, USA). If a significant difference was observed between the treated groups, Dunnett's test was applied. Differences with p<0.05 were deemed to indicate statistical significance.
RESULTS
Effects of paclitaxel and vinorelbine on the proliferation and viability of PDGF-BB-stimulated VSMCs via microtubule regulation
We carried out the MTT assay to test the effects of paclitaxel and vinorelbine on VSMC proliferation and viability via microtubule regulation (Figs. 1A and B). We also observed microtubules of VSMCs using immunofluorescence analysis (Fig. 1C). After binding to microtubules, paclitaxel enhances tubulin polymerization, resulting in stabilizing the microtubules and subsequently arresting metaphase of cell cycle [25]. Vinorelbine binds to free-tubulin dimers, and causes aggregation, resulting in the prevention of tubulin polymerization [26]. The concentration of these drugs used in this study were determined by performing preliminary tests for selecting doses at maximal effects without cytotoxicity (see Supplementary Fig. 1 of Supporting Information). Serum-starved VSMCs were pretreated with 1 µM paclitaxel, 0.2 µM vinorelbine, or 0.1% DMSO as a control for 24 h, and then the VSMCs were treated with or without 25 ng/ml PDGF-BB for 24–72 h. As shown in Fig. 1A, the proliferation of PDGF-BBstimulated VSMCs was inhibited significantly by paclitaxel and vinorelbine in a time-dependent manner.
Vinorelbine inhibited the proliferation of PDGF-BB-stimulated VSMCs by almost 100% at 2 days, and paclitaxel blocked the proliferation by almost 100% at 3 days. We also evaluated the cytotoxic effects of paclitaxel and vinorelbine. The viability of VSMCs was significantly decreased by vinorelbine at 2 days, and by paclitaxel and vinorelbine at 3 days. Paclitaxel led to inhibition of cell proliferation and induction of apoptosis via the stabilization of microtubules [27], and vinorelbine has demonstrated anti-proliferation and induction of apoptosis as a microtubuledestabilizing agent [28]. We confirmed the microtubule regulation of VSMCs by paclitaxel and vinorelbine through immunofluorescence staining of α-tubulin (component of microtubule). Fig. 1C shows that the microtubules were more likely to have a line shape in paclitaxel-treated VSMCs than in untreated VSMCs. However, vinorelbine-treated VSMCs were not observed to have a line shape; instead, microtubules were observed as dots. These results indicate that the proliferation of PDGF-BB-stimulated VSMC is inhibited by paclitaxel and vinorelbine via microtubule regulation. Additionally, VSMC viability was decreased by paclitaxel and vinorelbine.
Effects of microtubule regulation on caspase-3 in PDGF-BB-stimulated VSMCs
To examine the mechanism responsible for the apoptotic effects of paclitaxel and vinorelbine via microtubule regulation, we measured the cleavage of caspase-3 (from 35 to 17–19 kDa) as an apoptosis marker [22] and nuclear fragmentation [29], using western blotting and fluorescent staining of nuclei, respectively (Fig. 2). Serum-starved VSMCs were incubated with 1 µM paclitaxel, 0.2 µM vinorelbine, or 0.1% DMSO as a control for 24 h, and the VSMCs were incubated with or without 25 ng/ml PDGF-BB for 0–72 h. Fig. 2A shows that the PDGF-BB-stimulated VSMCs increased caspase-3 cleavage by microtubule regulation in a timedependent manner. The cleavage of caspase-3 through microtubule regulation in PDGF-BB-stimulated VSMCs was larger than that in unstimulated VSMCs. Next, we confirmed nuclear fragmentation, which is a feature of apoptosis [30]. Fig. 2B shows that the PDGF-BB-stimulated VSMCs demonstrated nuclear fragmentation by microtubule regulation. These results indicate that the microtubule-regulated VSMCs increasingly induce apoptosis by PDGF-BB stimulation through the blockade of microtubule dynamic formation.
Effects of autophagy via microtubule regulation and effects of caspase-3 cleavage via autophagy regulation in PDGF-BB-stimulated VSMCs
To investigate the effects of microtubule regulation on VSMC autophagy, we measured the autophagy activity of PDGF-BBstimulated VSMCs via microtubule regulation. 3-MA inhibits autophagy by the inhibition of type III phosphatidylinositol 3-kinases [31]. Activation of mechanistic target of rapamycin (mTOR) leads to suppression of autophagy via phosphorylation of multiple autophagy-related proteins such as ULK1 (UNC-5 like autophagy activating kinase 1), ATG13 (autophagy related gene 13), AMBRA1 (autophagy/beclin 1 regulator 1), and ATG14L (baclin 1-associated autophagy-related key regulator) [32]. Rapamycin promotes autophagy through inhibition of mTOR [33]. The concentrations of 3-MA and rapamycin in VSMC studies are widely used at 5 mM [34] and 0.2 µM [35], respectively. Thus, after confirming the effect of 5 mM 3-MA and 0.2 µM rapamycin on LC3-II levels (see Supplementary Fig. 2 in Supporting Information), these concentrations of 3-MA and rapamycin were used in this study.
The autophagy activity was evaluated by the conversion levels of LC3-I to -II [36] using western blotting (Fig. 3). Quiescent VSMCs were incubated with 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, 0.2 µM rapamycin, or 0.1% DMSO as a control for 24 h, and VSMCs were incubated with or without 25 ng/ml PDGF-BB for 0–48 h, and 0.1 µM bafilomycin A1 was treated 4 h before the end of the reaction. The PDGF-BB-stimulated or microtubule-regulated VSMCs increased autophagy activity in a time-dependent manner (Fig. 3A). Bafilomycin A1 is known to induce autophagosome accumulation through the inhibition of autophagosome and lysosome fusion [37]. Thus, bafilomycin A1 can be used to accurately measure LC3-II levels on the membrane of autophagosomes [36]. Figs. 3B and C show that VSMC autophagy activity increased microtubule regulation with PDGF-BB compared with that in unregulated microtubules or unstimulated VSMCs. The results were similar to those with the cleavage of caspase-3 shown in Fig. 2. We also confirmed the effects of viability via autophagy regulation in PDGF-BB-stimulated VSMCs. Fig. 3D shows that VSMCs inhibited autophagy by 3-MA (autophagy inhibitor), which increased caspase-3 cleavage. However, VSMCs promoted autophagy by rapamycin (autophagy stimulator), which decreased caspase-3 cleavage. These results suggest that autophagy plays an important role as a defense mechanism in inducing apoptosis by microtubule regulation of VSMCs.
Effects of the regulation of microtubules and autophagy on proliferation and viability in PDGF-BB-stimulated VSMCs
To further understand the role of autophagy in VSMC proliferation and viability, we investigated the regulatory effects of autophagy using 5 mM 3-MA and 0.2 µM rapamycin in microtubule-regulated VSMCs. The proliferation and viability of VSMCs were determined using the cell counting and MTT assays. Serum-starved VSMCs were cultured using 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, 0.2 µM rapamycin, or 0.1% DMSO as a control for 24 h, and the VSMCs were incubated with or without 25 ng/ml PDGF-BB for 48 h. Fig. 4A shows that the cell number of VSMCs was significantly more decreased by the regulation of microtubules and inhibition of autophagy than by only microtubule-regulated VSMCs. Moreover, activated VSMCs exhibited a decreased cell number via the regulation of microtubules and inhibition of autophagy compared with that of conditioned VSMCs not treated with PDGF-BB. As shown in Fig. 4B, the results of the inhibition of autophagy (Fig. 4A) and via the promotion of autophagy demonstrated the opposite pattern. Autophagy-promoted VSMCs significantly increased the viability and inhibited proliferation by rapamycin.
We further confirmed the role of autophagy using the MTT assay. The MTT assay is a method of evaluating cell proliferation and survival through mitochondrial activity [38]. Figs. 4C and D show that the VSMCs exhibited decreased proliferation and viability through the regulation of microtubules and inhibition of autophagy. However, the promotion of autophagy increased viability and inhibited proliferation in PDGF-BB-stimulated VSMCs. Taken together, microtubule regulation of PDGF-BB-stimulated VSMCs decreased cell viability via mitochondrial damage. Thus, activation of autophagy is a defense mechanism against the decrease in cell viability by microtubule regulation. Additionally, the proliferation was inhibited by almost 100% in conditioned VSMCs through autophagy promoting and microtubule regulation.
Intracellular ROS generation via the regulation of microtubules and autophagy in PDGF-BB-stimulated VSMCs
Generation of ROS induces apoptosis by the damage of VSMCs through oxidative stress [39]. To investigate whether intracellular ROS generation is involved in the regulation of microtubules and autophagy in PDGF-BB-stimulated VSMCs, intracellular ROS levels were measured using the H2DCFDA assay (Fig. 5). Quiescent VSMCs were pretreated with 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, 0.2 µM rapamycin, or 0.1% DMSO as a control, followed by incubation for 24 h, and then the VSMCs were incubated with or without 25 ng/ml PDGF-BB for 48 h. Fig. 5A shows that the VSMCs exhibited increased intracellular ROS generation by microtubule regulation. The ROS levels revealed significantly increased regulation of microtubule-regulated and autophagy-inhibited VSMCs compared with that in only microtubule-regulated VSMCs, whereas the ROS levels of autophagystimulated VSMCs were significantly decreased compared with that in only microtubule-regulated VSMCs. Intracellular ROS were increasingly generated in PDGF-BB-stimulated and microtubule-regulated VSMCs (Fig. 5B). The PDGF-BB-stimulated VSMCs also demonstrated changes in the ROS level with the same pattern as shown in Fig. 5A through regulation of autophagy (Fig. 5B). These results indicate that the microtubule-regulated and PDGF-BB-stimulated VSMCs exhibited increased ROS generation, and the promotion of autophagy decreased intracellular ROS generation.
Effects of the regulation of intracellular ROS, microtubules and autophagy on proliferation and viability in PDGF-BB-stimulated VSMCs
To determine the effects of intracellular ROS regulation on the regulation of microtubules and autophagy in PDGF-BB-stimulated VSMCs, the ROS level, proliferation, and viability were measured using H2DCFDA, cell counting, and MTT assays, respectively. Serum-starved VSMCs were cultured with 1 µM paclitaxel, 0.2 µM vinorelbine, 5 mM 3-MA, 0.2 µM rapamycin, 5 mM NAC, or 0.1% DMSO as a control for 24 h, and VSMCs were incubated with or without 25 ng/ml PDGF-BB for 48 h. As shown in Fig. 6A, 5 mM NAC had no toxic effects on VSMCs after 48–72 h. The microtubule- and autophagy-regulated and PDGF-BB-stimulated VSMCs inhibited intracellular ROS generation by 5 mM NAC (Fig. 6B). Fig. 6C shows that the microtubule-regulated VSMCs inhibited autophagy, causing a decreased cell number. However, the decrease in cell number was significantly alleviated in NAC-treated VSMCs. The reduction in the cell number of microtubule-regulated and PDGF-BB-stimulated VSMCs was also significantly alleviated in the NAC-treated groups (Fig. 6D). The cell numbers of microtubule-regulated and autophagy-stimulated VSMCs were similar to those of the NAC-treated groups (Figs. 6E and F). Together, these results suggest that the reduction of ROS by activating autophagy can enhance the VSMC viability.
DISCUSSION
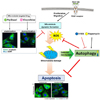
The present study yielded three major findings: (1) the potent cytotoxic effect is induced in PDGF-BB-stimulated VSMCs through excessive generation of intracellular ROS by blocking microtubule dynamic formation; (2) apoptosis is increased more in autophagy-inhibited and conditioned VSMCs than in only microtubule-regulated VSMCs; and (3) the promotion of autophagy decreases apoptosis via the reduction of ROS levels. These findings are illustrated in Fig. 7.
The anti-cancer drugs paclitaxel and vinorelbine stabilize and destabilize microtubules, respectively, inducing the inhibition of cell proliferation and migration and induction of apoptosis [4041]. Paclitaxel and vinorelbine decreased the proliferation and viability levels of VSMCs in a time-dependent manner (Figs. 1A and B). It was also confirmed that the drugs inhibit the dynamic formation of microtubules (Fig. 1C). Therefore, we confirmed that paclitaxel and vinorelbine inhibit the proliferation of VSMCs by microtubule regulation, supporting the results of previous studies [40]. Caspase-3 is a major marker of apoptosis and promotes the apoptosis pathway [42]. We observed that microtubule dynamic formation in VSMCs was suppressed using paclitaxel and vinorelbine, and stimulation with PDGF-BB induced apoptosis via the cleavage of caspase-3 (Fig. 2). PDGF-BB-stimulated VSMCs activate microtubule dynamic formation, as well as promote proliferation and migration [43]. Suppression of microtubule dynamic formation may induce the apoptosis pathway of activated VSMCs.
Because the autophagy activity of PDGF-BB-stimulated VSMCs is increased and plays an important role in resistance to oxidative stress [36], we further evaluated the effects of microtubule regulation on PDGF-BB-stimulated VSMC autophagy, as well as the apoptotic effects of autophagy regulation on VSMCs. The autophagy levels of microtubule-regulated VSMCs were increased in a time-dependent manner. The autophagy levels of microtubule-regulated and PDGF-BB-stimulated VSMCs were further increased (Figs. 3A, B and C). Additionally, inhibition of autophagy using 3-MA increased apoptosis, and activation of autophagy using rapamycin maintained the viability (Fig. 3D). These results indicate that the control of apoptosis by autophagy regulation in VSMCs is possible.
Apoptosis was induced by the regulation of microtubules and stimulation of PDGF-BB (Fig. 2), and autophagy activity was also increased in the same conditioned VSMCs (Fig. 3A). Previous studies have reported that the increased activity of autophagy inhibits the apoptosis pathway [4445]. Beclin 1 plays an important role in autophagy. Beclin 1 forms core complexes (beclin 1-vps34-vps15), that induce the autophagy pathway, and it also plays an anti-apoptotic role under some conditions, such as chemotherapy, irradiation, immunotherapy, nutrient deprivation, and angiogenesis [46]. Therefore, it may be considered that the autophagy pathway is activated as a defense mechanism of conditioned VSMCs in apoptosis.
We further confirmed the effects of autophagy on the proliferation and viability of conditioned VSMCs. 3-MA significantly reduced the number of VSMCs through autophagy inhibition. The proliferation of VSMCs was inhibited by autophagy stimulation using rapamycin, but the viability was maintained (Figs. 4A and B). Since MTT assays have revealed cell viability and proliferation in response to mitochondrial activity [38], the low values of the MTT assay indicate low levels of mitochondria activity and mitochondrial dysfunction. The results shown in Fig. 4C and D revealed the same patterns as those shown in Figs. 4A and B. These results suggest that apoptosis is induced by mitochondrial damage in microtubule-regulated and PDGF-BB-stimulated VSMCs, and that mitochondrial damage is decreased by promoting autophagy.
Oxidative stress due to excessive ROS levels damages cellular organelles and induces apoptosis [4748]. PDGF-BB-stimulated VSMCs exhibited increased ROS, which acts as a signaling pathway molecule [49]. Antioxidants reduce PDGF β-receptor phosphorylation through the inhibitory oxidation of protein tyrosine phosphatases [50]. The ROS levels of microtubule-regulated VSMCs were substantially increased (Fig. 5A). The ROS levels of microtubule-regulated and PDGF-BB-stimulated VSMCs were higher than those of only microtubule-regulated VSMCs (Fig. 5B). The increase in autophagy activity affects ROS reduction [51]. Promoting the autophagy of VSMCs decreased the ROS levels (Fig. 5). Thus, these results suggest that the promotion of autophagy in the conditioned VSMCs decreases the ROS levels and prevents cell damage. We also confirmed the relationship between ROS and autophagy. ROS levels were reduced using NAC to suppress oxidative stress. Conditioned VSMCs were incubated with NAC, improving the viability of the VSMCs (Figs. 6C and D). Additionally, the anti-apoptotic effects of the autophagy stimulator and effects of NAC on VSMC viability were similar (Figs. 6E and F). These results suggest that the inhibition of oxidative stress through ROS reduction plays an important role in the viability of microtubule-regulated and PDGF-BB-stimulated VSMCs, and the promotion of autophagy is thought to maintain the viability of cells through ROS reduction.
In conclusion, our results indicate that the increased levels of autophagy in the microtubule-regulated and PDGF-BB-stimulated VSMCs reduce the levels of intracellular ROS and suppress mitochondrial damage. Therefore, we suggest that regulation of the autophagy level in VSMCs as a tool for the treatment of diseases, such as arteriosclerosis and vascular restenosis, is a good strategy to promote autophagy.






 XML Download
XML Download