

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Before the discovery of direct activation and interaction of Gβγ with the potassium channel in the heart [1], the Gα subunit was considered the only effector, and the Gβγ complex was considered a signal inhibitor molecule. However, after the discovery of several biological effects of Gβγ, many studies have attempted to understand its functions. It is currently known that the Gβγ complex has its own properties and functional roles in cellular signaling pathways [23]. Gβγ has specific tissue expression patterns, and specific coupling between the GPCRs [3]. For example, a study showed that Gβ1γ1 assisted Gαt binding to rhodopsin, but Gβ1γ2 did not [4]. Gα13β1γ3 complexes are coupled with the angiotensin receptor (AT1A) in rat portal vein myocytes, and enhance cytosolic calcium ion transport [5]. These previous studies as well as others suggest that each component of the Gβγ subunit complex has its own unique role in specific tissues.
Interestingly, recent genetic studies have reported that mutations of GNB5 (the gene encoding Gβ5) were associated with the symptoms of sinus bradycardia, cognitive disability [6], and attention deficit hyperactivity disorder (ADHD) [78] in the human. Moreover, a mouse model that lacked Gβ5 showed hyperactivity [9].
GNB5 related symptom related with calcium signaling. Calcium signaling is closely related with cardiac and neural function. In particular, store-operated calcium entry (SOCE) plays important roles in the heart [101112]. SOCE allows replenishment of sarcoplasmic reticulum calcium in the cardiomyocytes; therefore, defects in SOCE lead to some cardiac diseases [1213]. In neurons, calcium signaling is also important. Spontaneously hypertensive rats, as an animal model of ADHD [14], have a dysfunction of calcium signaling [1516] and lower brain Ca2+-ATPase activity [17].
Consequently, previous studies have implied a relationship of the GNB5 gene and SOCE. However, there are no studies describing this relationship, and therefore we investigated the regulatory mechanism of GNB5-induced effects on SOCE.
Go to : 
METHODS
cDNA and transfection
The genes, GNB1 (NCBI accession number: NM_002074), GNB5 (NCBI accession number: NM_006578), GNG11 (NCBI accession number: NM_004126), and GNG13 (NCBI accession number: NM_016541), were cloned into the mammalian expression vectors, pEGFP-N1 and p3XFLAG-CMV14. The mutant Orai1R91W [1819] and ΔERM-Stim1D76A [2021] were also used. All the samples were incubated for 24 h after transfection using Lipofectamine 2,000 (Invitrogen, Carlsbad, CA, USA).
Cell culture
HEK293T cells were maintained in high-glucose Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum and 1× antibiotic-antimycotic reagent (Gibco, Gaithersburg, MD, USA) in 95% CO2, 5% O2, at 37℃. Subculturing was done with trypsin/EDTA when the cells reached confluency. The cells used in the calcium imaging experiments were cultured on 24 mm×24 mm glass slides.
Measurement of intracellular calcium
The cells were cultured on 24 mm×24 mm glass slides at a density of 1.5×106 cells/ml for 48 h and then treated with 4 µM Fura-2/AM and 0.05% pluronic acid F-127 for 30 min in physiological salt solution at room temperature. Fura-2/AM fluorescence was measured at an excitation wavelength of 340/380 nm, and an emission wavelength of 510 nm (expressed as the ratio=F340/F380), using an imaging system (Molecular Devices, Sunnyvale, CA, USA). The emitted fluorescence was monitored using a CCD camera (CoolSNAP, Tucson, AZ, USA) attached to an inverted microscope. Fluorescence images were obtained at 2-s intervals. All data were analyzed using MetaFluor software (Molecular Devices, Downingtown, PA, USA). Intracellular calcium is shown as representative traces. The data represent the mean±S.E.M. ΔPlateau was defined as the value that difference of the Fura-2/AM ratio between the injection point of 2 mM Ca2+ solution and highest value.
Statistical analysis
The statistical significance of differences between groups was determined using the paired Student's t-test. In all cases, p values< 0.05 were considered significant.
Go to :
RESULTS
GNB5 expression increases SOCE
To investigate the role of the Gβ and Gγ subunits in SOCE, we overexpressed GNB1, GNB5, GNG11, and GNG12 in HEK293T cells. After the expression of each protein subunit was confirmed, we measured the amount of SOCE (Fig. 1A). To maximize SOCE, we used 25 µM cyclopiazonic acid (CPA), and induced calcium entry using 2 mM Ca2+ physiological salt solution. SOCE was increased after exogenous overexpression of the GNB5 gene (Fig. 1B), suggesting that Gβ5 protein affects the SOCE in HEK293T cells.
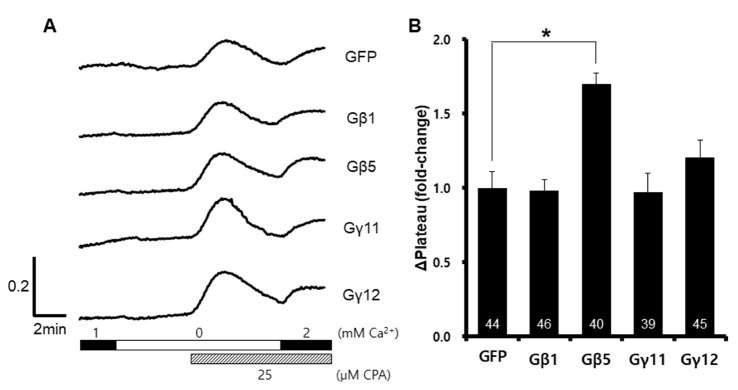 | Fig. 1Enhanced store-operated Ca2+ entry induced by overexpression of GNB5After depletion of ER Ca2+ stores using cyclopiazonic acid and EGTA, Ca2+ entry was measure by Fura2-AM fluorescent signal ratios. A representative trace (A) summarizes the data (B), and the number inside the column is the total cell number from multiple independent experiments (n=3-4). Data were expressed as the mean±SEM. *p<0.05 compared with GFP.
|
GNB5 enhances Stim1-Orai1 dependent SOCE
Because Stim1-Orai1 complexes are known as ubiquitous store-operated Ca2+ channels [22], we investigate SOCE after co-expression of Stim1, Orai1, and Gβ5. SOCE was significantly increased when all the components were expressed (Fig. 2B). With Gβ5 expression, the SOCE was approximately 2.5- fold higher than that with Stim1 or Orai1 expression. SOCE was also enhanced in Stim1-Gβ5 expressing cells and in Orai1-Gβ5 expressing cells. These results suggested that Gβ5 interacts with Orai1 or Stim1, or both.
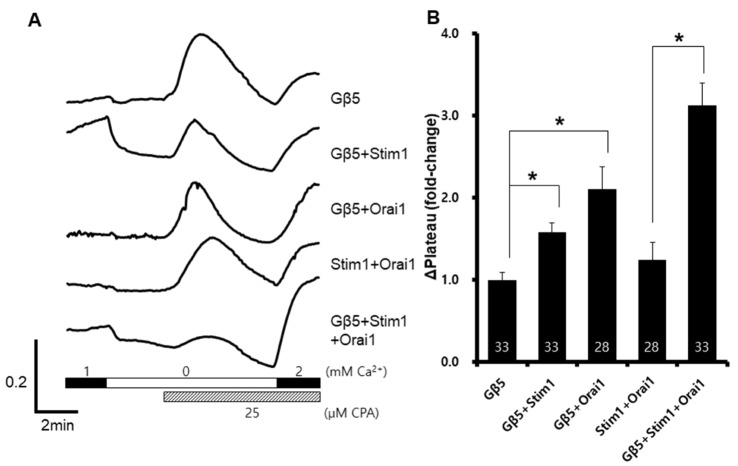 | Fig. 2Increased Stim1-Orai1 dependent store-operated Ca2+ entry after GNB5 overexpression.Gβ5, Stim1, and Orai1 were co-expressed in HEK293T cells. A representative trace (A) summarizes the data (B), and the number inside the column is the total cell number from multiple independent experiments (n=3-4). Data were expressed as the mean±SEM. *p<0.05 compared with control.
|
Mutant Orai1 has no effect on the GNB5-induced enhancement of SOCE
To confirm the target of Gβ5, we co-expressed Orai1R91W, a loss-of-function mutant of Orai1, with Gβ5. Although mutant Orai1 was expressed, SOCE was still enhanced by Gβ5 expression (Fig. 3B). This might be a result of the presence of endogenous Orai1 and its participation in SOCE.
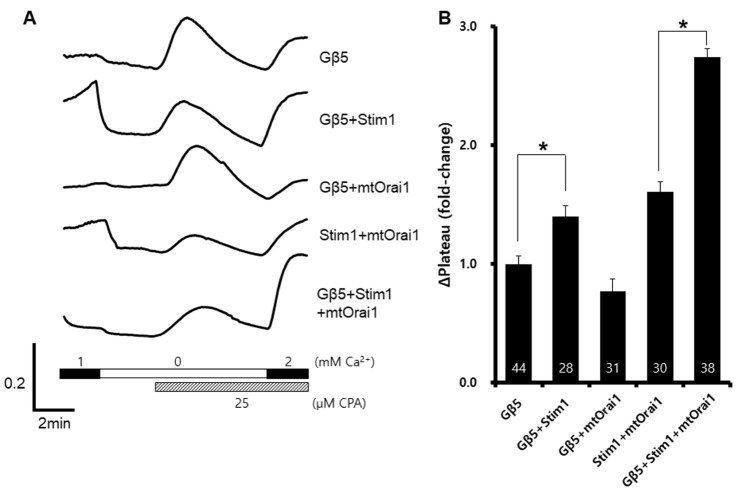 | Fig. 3Effects of a loss-of-functional Orai1 mutant on store-operated Ca2+ entry.Wild-type Gβ5, wild-type Stim1, and Orai1R91W were co-expressed in HEK293T cells. A representative trace (A) summarizes the data (B), and the number inside the column is the total cell number from multiple independent experiments (n=3-4). Data were expressed as the mean±SEM. *p<0.05 compared with control.
|
Stim1-ΔERM abolishes the augmentation of SOCE induced by GNB5 gene expression
ΔERM-Stim1D76A is a dominant-negative form of STIM1 (DN-STIM1). When we expressed DN-STIM1 with GNB5, the enhancement of SOCE was diminished (Fig. 4B). This result supported the hypothesis that GNB5 may interact with STIM1.
 | Fig. 4Diminished enhancement of the store-operated Ca2+ entry induced by Gβ5 and the co-expression of a dominant-negative Stim1.Wild-type Gβ5, ΔERM-Stim1D76A, and wild-type Orai1 were co-expressed in HEK293T cells. A representative trace (A) summarizes the data (B), and the number inside the column is the total cell number from multiple independent experiments (n=3-4). N. D.=not determined.
|
Go to :
DISCUSSION
GPCR signaling is a well-established pathway by which intra-cellular signals interact with extracellular signals [23]. Coupling between phospholipase Cβ (PLCβ) induces inositol-1, 4, 5-triphosphate (IP3) generation and subsequent cytosolic Ca2+ ion cascades [24]. Specific Gβγ subunit complexes activate PLCβ and cause its translocation to the cytosol, to subsequently interact with the IP3 receptor in the endoplasmic reticulum (ER), and induce changes in Ca2+ conductance [2526]. Evidence also suggests a direct activation of IP3 by Gβγ complexes [27]. However, the role of Gβγ complexes in SOCE remains unknown. From our present results, we concluded that Gβ5 expression enhanced SOCE (Fig. 1B).
The mechanism by which Gβ5 increased SOCE remains unknown. Stim1-Orai1 dependent SOCE was significantly increased by the expression of Gβ5 (Fig. 2B). The replacement of wild-type Orai1 with a loss-of-function Orai1 mutant, Orai1R91W, did not alter SOCE. However, a loss-of-function Stim1 mutant, ΔERM-Stim1D76A, inhibited the SOCE induced by Gβ5. These results suggested that GNB5 gene expression may affect Stim1 action, however, whether this is a direct or indirect relationship is unclear. Further studies are needed to evaluate the interactions of Gβ5 with Stim1.
The Stim1 protein has an EF-hand domain for calcium sensing at the ER lumen [28]. The cytosolic portion of Stim1 begins with an ERM domain that starts with a coiled-coil domain for oligomerization and oligomer-induced recruitment of Orai1 protein to the plasma membrane [19]. In the ERM domain of the Stim1 protein, the Stim1 Orai1 activating region (SOAR) performs key functions in the activation of Orai1 [1929]. In our study, ΔERM-Stim1D76A inhibited the SOCE induced by Gβ5. Gβ5 also has α-helical extension of 20 amino acids forms a coiled-coil domain [30]. This suggests that the coiled-coil domain may enhance the recruitment of the Orai1 protein.
Many Stim1-modulating molecules have been identified. For example, SARAF and TEM66 bind to the Stim1 SOAR domain to cause reduced Orai1 activity [31]. STIMATE and TEM110 interact with Stim1 to promote its conformational switch changes [32]. Cracm1, calcium related activating current modulator 1, is a plasma membrane protein that is essential for SOCE [33]. The R7 RGS protein, regulator of G-protein signaling protein, mediates Gβ5 neuronal signaling [89]. Similarly, GNB5 may act by modulating molecules related to STIM1.
The TRPC pathway is another important pathway of SOCE control in mammalian cells [192934]. In a mouse model that lacks Trpc3, reduced SOCE in pancreatic acinar cells and reduced amylase secretion were observed [35]. Stim1 SOAR domains are also important for activation of Trpc3 [19]. Previous studies indicated that Gβ5 interacts with Stim1, and its expression promotes Trpc3-dependent SOCE. Studies of SOCE in Gβ5-Trpc3 expressing cells, compared with cells expressing Trpc3 alone, have not been reported. Therefore, the role of Gβ5 and Stim1 should be further studied.
The mechanism and function of the Gβγ complexes still remain to be determined. Gβγ complexes are potential therapeutic targets, therefore, our findings may contribute to the development of novel approaches for cures of diseases.
Go to :
 XML Download
XML Download