

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hyperglycemia accelerates the reaction between glucose and proteins and promotes the formation of advanced glycation end products (AGEs). Interacting with its specific receptor, RAGE, AGEs form cross-links with many macromolecules such as collagen [12]. AGE-related cross-linking of collagen is associated with aortic wall matrix stiffness and AGEs stimulates the release of profibrotic growth factors such as transforming growth factor (TGF)-beta, promote collagen deposition, increase inflammation, and ultimately lead to tissue fibrosis [345]. It has been suggested that connective tissue growth factor (CTGF) is a potent inducer of extracellular matrix (ECM) in diabetes.
CTGF is a widely known as a hallmark of fibrosis in multiple that is implicated in fibroblast proliferation, cellular adhesion, angiogenesis, and ECM synthesis [6]. It has been reported that CTGF promotes vascular smooth muscle cell (VSMC) proliferation, migration, and production of ECM, which may play a role in the development and progression of atherosclerosis [7]. The addition of CTGF to primary mesangial cells induced fibronectin production, cell migration, and cytoskeletal rearrangement which were associated with recruitment of Src and phosphorylation of p42/44 MAPK and protein kinase B [8]. It has been reported that high glucose induces CTGF expression and ECM accumulation in VSMCs via inactivation of ERK1/2 [9].
Statins are a class of drugs used to lower cholesterol levels by inhibiting the enzyme HMG-CoA reductase, which plays an important role in the production of cholesterol in liver [10]. It has been demonstrated that statins have significant immunomodulatory effects and reduce vascular injury [11]. A previous study by our research team showed that pitavastatin inhibited the effects of angiostetin II (AngII)-induced insulin-like growth factor binding protein (IGFBP)-5, namely, VSMC proliferation, migration, and ECM accumulation [12]. The mechanisms of hyperglycemiainduced vascular remodeling are incompletely understood; however, metabolic and mechanical factors seem to play important roles. With this background, we examined whether AGEs induced ERK1/2 and JNK signaling pathways, which could result in the stimulation of CTGF secretion and increase in VSMC proliferation and migration, thereby resulting in ECM accumulation. We observed that these effects were reversed by administration of fluvastatin. Our results demonstrated the inhibitory effect of fluvastatin on AGE-induced VSMC proliferation, migration, and production of ECM, which were associated with secretion of CTGF.
METHODS
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and antibiotics (penicillin and streptomycin) were purchased from Gibco BRL (Rockville, MD, USA). Anti-β-actin, 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-CTGF, anti-JNK, anti-pJNK, anti-p38, anti-pp38, anti-ERK1/2, anti-pERK1/2, goat anti-rabbit IgG, goat anti-mouse IgG, donkey anti-goat IgG, enhanced chemiluminescence (ECL), Egr-1, and control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). AGE-BSA was obtained from Calbiochem (Darmastadt, Germany), PD98059, U0126, SP600125 and SB203580 were purchased from Calbiochem (San Diego, CA, USA). Lipofectamine2000TM was purchased from Invitrogen (Carlsbad, CA, USA). Recombinant CTGF was purchased from R&D system (Minneapolis, ME, USA) and α-tubulin from Sigma (St. Louis, MO).
Primary cell culture
Sprague-Dawley rats were anesthetized with pentobarbital (50 mg/kg). The thoracic aorta removed, cleaned of adhering fat and connective tissue, and thoracic aortic rings were prepared. The vascular endothelium was mechanically removed by rubbing gently with wooden stick to remove endothelium. VSMCs were processed using a 1 mm chop setting in a 100 mm culture dish, and cultured with 50% FBS-DMEM with 1% antibiotics at 37℃ in a humidified atmosphere containing 5% CO2 for 7 days. VSMCs were maintained in DMEM with 10% FBS and 1% antibiotic (penicillin 10,000 U/ml, streptomycin 10,000 µg/ml). We used VSMCs from passages 4 to 8 at 70–90% confluence in 10 cm dishes, and cell growth was arrested by incubation of the cells in serum-free DMEM for 24 h prior to use.
Transfection of siRNA
The cells were transfected with siRNA using Lipofectamine 2,000 reagent, according to the manufacturer's instructions. Aliquots of 1×104 cells were plated on 60-mm dishes 1 day before transfection, and grown to approximately 70% confluence. The cells were then transfected with siRNA (20 nM Egr-1, 20 nM control) and 3 µl of lipofectamine for 6 h in Opti-MEM®I reduced serum medium (Invitrogen), followed by incubation for 48 h.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated by TRIzol, and a reverse transcription reaction was conducted using TaqMan reverse transcription reagents, following the manufacturer's instructions. RT-PCR was conducted with 1 µl of template cDNA and power SYBR Green in ABI PRISM 7500. Quantification was performed using the efficiency-corrected ΔΔCq method. Primers used to amplify DNA sequences were as follows: CTGF, forward 5′-TCAGGCACCCTCATATAATC-3′ and reverse 5′-GACAATAGTCCACACCAGA-3′,
Fibronection, forward 5′-TTACTATGGGATGGGGTCCA-3′ and reverse 5′-TGCCAAAACTGTTCACCAAA-3′, collagen I, forward 5′-AAACCACCCTGAAAGCACAG-3′ and reverse 5′-AGTGTTCTGGTGATGCCACA-3′; and collagen III, forward 5′-GGAGCCAAAAGGGTCATCAT-3′ and reverse 5′-GTGATGGCATGGACTGTGGT-3′, GAPDH, forward 5′-AGGGAAATCGTCGTGCGTGAG-3′ and reverse 5′-CGCTCATTGCCGATAGT-3′. The PCR conditions were as follows: preliminary denaturation at 50℃ for 2 min; 95℃ for 10 min, 95℃ for 15 s, and 60℃ for 1 min.
Western blotting
The cells were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with 1 mmol/L phenylmethylsulfonyl fluoride (PMSF) and 0.01 mmol/L protease inhibitor cocktail (PIC), and incubated on ice for 15 min, followed by centrifugation at 15,000 × g for 10 min at 4℃. Protein concentration was determined from the supernatant obtained by centrifugation using the Bradford assay. Proteins were separated by SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membranes were immunoblotted with primary antibodies (1:1000), followed by immunoblotting with corresponding secondary antibodies (1:5000). Signals were visualized by using electrochemiluminescence (ECL) detection reagents (Millipore, Temecula, CA), according to the manufacturer's instructions.
MTT assay
The cells were cultured on 24-well plates. When the cells are approximately 80% confluent, the medium was replaced with serum free DMEM. After starvation, the cells were pretreatment with or without fluvastatin (5 µM) and then stimulated with AGEs (10 µg/ml) for 24 h. MTT reagent was added and incubated for 4 h at 37℃, and then washed with PBS and eluted by adding dimethyl sulfoxide (DMSO). Cell proliferation was measured using microplate reader (Biorad) at 570 nm.
Wound scratch assays
When cells culltured to confluence or near >90% in either 6-well dishes. The medium was replaced with serum free DMEM, and the cells were incubated overnight. Using a sterile 200 µl pipet tip, one separate wound was scratched through the cells moving perpendicular to the line drawn in the step above. After scratching, the cells were pretreated with fluvastatin in the presence or absence of recombinant CTGF (100 nM) for 1 h and then exposed to AGEs (10 µg/ml) for 36 h. The images were acquired using a phase contrast microscope (40×).
Flow cytometric analysis
The cells (1×105) were trypsinized and fixed in 95% ethanol overnight. Fixed cells were stained with propidium iodide (PI; 50 µg/ml) for 30 min at 37℃. PI enters the cells and stains the nucleus. PI-stained cells were filtered using a 5 mL polystyrene round bottom tube with a cell-strainer cap prior to flow cytometry. All flow cytometry measurements were done using a FACSCalibur (Becton Dickinson, San Jose, CA, USA). Cell cycle analysis was performed using Cell-Quest Pro-Software.
RESULTS
Fluvastatin inhibits AGE-induced CTGF expression in VSMCs
To determine whether AGEs induce CTGF expression in VSMCs, the cells were treated with 10 µg/ml AGEs at various times (0, 6, 12, 18, and 24 h). AGEs increased CTGF mRNA level in a time dependent manner, and this effect peaked at 24 h (Fig. 1A). AGE-induced CTGF protein level peaked at 12 h, and then decreased slightly (Fig. 1B). Next, we examined the effect of fluvastatin on AGE-induced CTGF expression. Treatment with 5 µM fluvastatin dramatically inhibited mRNA and protein levels of CTGF (Figs. 1C and D). These results indicated that AGE-induced CTGF mRNA and protein expression was inhibited by fluvastatin treatment.
AGEs induce CTGF expression in VSMCs via ERK/JNK/Egr-1 pathways
To determine the signaling mechanism involved in the induction of CTGF expression by AGE, we first investigated the role of mitogen-activated protein kinase (MAPK) on AGE-induced CTGF in VSMCs. AGEs increased p-ERK1/2, p-JNK and p-p38 expression in a time-dependent manner (Fig. 2A). To examine the role of MAPK, we treated the cells with MAPK-specific inhibitors, MEK1/2 inhibitors PD98059 or U0126, JNK inhibitor SP600125, and p38 MAPK SB203580 on AGE-induced CTGF protein expression and proliferation in VSMCs. We observed that AGE-induced CTGF expression and proliferation in VSMCs by regulating ERK1/2 and JNK inhibitors, but not by the p38 inhibitor (Figs. 2B and C). These results demonstrate that AGE induced CTGF expression and proliferation in VSMCs by regulating ERK1/2 and JNK signaling mechanism. It has been suggested that Egr-1 is a downstream of ERK1/2 MAPK pathway [13]; therefore, we examined the effect of ERK1/2 on Egr-1 expression and VSMC proliferation. To block ERK1/2 MAPK actions, we used MEK1/2 inhibitors, PD98059 or U0126. MEK1/2 inhibitors efficiently inhibited Egr-1 expression in AGE-induced VSMCs (Fig. 3A). Next, to determine the role of Egr-1 on CTGF expression, we blocked Egr-1 using adenoviral-delivered Egr-1 siRNA. Knockdown with Egr-1 suppressed CTGF expression and proliferation in AGE-treated VSMCs (Figs. 3B and C). These results indicated that Egr-1 is a key factor in AGE-induced CTGF expression and VSMC proliferation. Together, our results suggested that AGE induces CTGF and VSMC proliferation via the ERK/JNK/Egr-1 pathway. We also observed that fluvastatin suppressed AGE-induced ERK1/2, JNK and Egr-1 expression in VSMCs (Figs. 2D and 3D).
Fluvastatin inhibits AGE-induced VSMC proliferation, migration and ECM production via inhibiting CTGF
To confirm whether fluvastatin affects VSMC proliferation induced by AGE, VSMCs were treated with AGE in presence or absence of fluvastatin, followed by MTT assay. As shown in Fig. 3A, the proliferative effect of AGE was significantly suppressed by fluvastatin. To determine the effect of fluvastatin on the production of ECM molecules, reverse transcriptase PCR was applied to test the mRNA level of fibronectin, collagen I and III. As shown in Figs. 4B-D, AGE significantly induced mRNA level of ECM molecules, fibronectin, collagen I and III, and the production of ECM molecules was significantly suppressed by fluvastatin. Notably, fluvastatin-mediated suppression of AGE-induced cell proliferation and ECM production were restored treatment with CTGF recombinant protein (Figs. 4B-D). In addition, to degermine the effect of fluvastatin on AGE-induced VSMC migration, wound scratch assay was performed to detect endogenous cell migration. As shown in Fig. 3E, fluvastatin significantly attenuated AGE-induced cell migration in response to wound injury, which was restored treatment with CTGF recombinant protein. Taken together, our findings suggest that fluvastatin regulates AGE-induced VSMC proliferation, migration and ECM production via CTGF dependent mechanism.
Fluvastatin increases cell cycle arrest through inhibition of CTGF in AGE-induced VSMCs
Cellular proliferation is regulated by the cell cycle consists of four distinct sequential phases: G0/G1, S, G2, and M. After vascular injury, VSMCs are stimulated to divide in response to mitogens, and they exit G1 phase and enter S phase. To examine the effects of fluvastatin on cell cycle, analyses were performed in AGE-induced VSMCs. As shown in Fig. 5, AGEs significantly blocked the cells in G0/G1 phase compared to the control, and fluvastatin significantly increased the cells in G0/G1 phase compared to AGE-treated. Next, we determined the role of CTGF in the modulation of AGE-induced cells in G1/G0 phase by fluvastatin. Fluvastatin-induced anti-proliferative action was repressed by treatment with CTGF recombinant protein (Figs. 5A and B), suggesting that fluvastatin blocks VSMC proliferation via CTGF, resulting in cell cycle arrest. To determine whether fluvastatin affected the cell cycle progression by modulating the expression of cell cycle regulatory genes including cyclin D, Cdk4, P27 and p21, we evaluated whether AGEs increased the levels of cyclin D and Cdk4 proteins and reduced p27 and p21 levels. Fluvastatin suppressed cyclin D1 and CDK4 protein levels, but enhanced p27 and p21 levels in VSMCs, and these effects were prevented by treatment with recombinant CTGF (Fig. 5C). These results indicated that fluvastatin ameliorated AGE-induced proliferation by inducing cell cycle arrest in VSMCs via modulation of CTGF expression.
DISCUSSION
Some studies have shown that AGEs may mediate diabetic vascular complications by stimulating ECM production. ECM accumulation in the cardiovascular system is involved in intimal plague formation in the atherosclerotic lesions in diabetic vessels [141516]. It has been recently demonstrated AGEs play a key role in neointimal formation after vascular injury [1718]. In fact, AGE-induced VSMC proliferation and ECM production are emerging as important mechanisms of atherosclerosis. In our previous study, we demonstrated that high glucose increases the levels of CTGF, resulting in the induction of VSMC proliferation, migration and ECM production [9]. Several studies revealed that CTGF activation markedly increased cell proliferation in normal cells as well as in tumor cells and VSMCs [19202122]. In the present study, we demonstrated that AGEs increases the expression of CTGF mRNA and protein (Figs. 1A and B), which resulted in the induction of VSMC proliferation, migration and ECM accumulation (Fig. 4). Yoon et al., demonstrated that simvastatin inhibits AGE-induced VSMC proliferation and neointimal formation after balloon injury in diabetic rats [17]. We determined that fluvastatin has a modulatory effect on AGE-induced CTGF expression. AGE-induced CTGF expression was significantly inhibited by treatment with fluvastatin (Figs. 1C and D). In addition, AGE-induced VSMC proliferation, migration and ECM accumulation were significantly suppressed by fluvastatin (Fig. 4). Therefore, we hypothesize that CTGF might influence AGE-induced fibrogenic progression in VSMCs. As shown in Fig. 4, the inhibitory effects of fluvastatin on AGE-induced VSMC proliferation, migration and ECM production were restored by treatment with CTGF recombinant protein. These finding imply that activation of CTGF signaling pathways may be a useful strategy to prevent diabetic vasculopathy.
Increased levels of AGEs result in an increased level of CTGF which has been shown to activate signaling cascades that activate MAPKs, including ERK1/2 and JNK that are implicated in VSMC proliferation, migration, and fibrosis [232425]. It has been reported that the ERK1/2 MAPK pathway is responsible for AGE-induced early activation of CTGF expression in VSMCs [26]. In this study, AGE-induced CTGF was inhibited by ERK1/2 inhibitors and JNK inhibitor but not p38 inhibitor (Fig. 2B). Egr-1 transcription factor, an ERK1/2 downstream target encoded by the immediate gene Egr-1, has been shown to orchestrate multiple events, leading to vessel remodeling [2728]. The major finding of this study is that AGE increased Egr-1 levels in VSMCs (Fig. 3A). To determine whether ERK1/2 MAPK regulates Egr-1 expression, we treated the cells with MEK1/2 inhibitors, PD98059 or U0126. It was observed the MEK1/2 inhibitors efficiently inhibited Egr-1 expression in AGE-stimulated VSMCs (Fig. 3B). In addition, we confirmed that knockdown of Egr-1 prevents CTGF induction and VSMC proliferation in AGE-stimulated VSMCs (Fig. 3C). Therefore, we demonstrated that AGE induced CTGF expression and proliferation in VSMCs by regulating ERK1/2, JNK, and Egr-1 signaling mechanism. Moreover, AGE-induced pERK1/2, pJNK, and Egr-1 expression was significantly attenuated by fluvastatin (Figs. 2D and 3D).
Before commencing the present study, we hypothesized that fluvastatin might reduce AGE-induced proliferation, and thus, we sought to identify the mechanism underlying the involvement of fluvastatin in AGE-induced cellular signaling. Treatment with AGE decreased cell cycle arrest at G0/G1 phase; however, these effects of AGE were lost when CTGF expression was depleted by CTGF siRNA (Fig. 5A). Some reports have shown that fluvastatin affected cell cycle progression by modulating the expression of cell cycle regulatory genes including cyclin D, Cdk4, P27 and p21 [29]. Treatment with AGE increased the protein levels of the proliferation markers, cyclin D1 and CDK4, and markedly decreased p21 and p27 levels. These effects of AGEs were recovered upon treatment with CTGF recombinant protein (Fig. 5C).
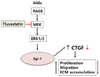
In conclusion, our current study we observed that ECM production, proliferation, and migration induced by AGE in VSMCs led to the induction of CTGF expression. Furthermore, we identified ERK1/2 and JNK activation and Egr-1 expression as mechanisms mediating AGE-induced CTGF expression (Fig. 6). Our findings provide new insights to understanding the molecular mechanism of CTGF-dependent fibrosis in diabetic vascular complication. In addition, fluvastatin therapy may allow better therapeutic application and foster the early use of statins in acute vascular disease.





 XML Download
XML Download