

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Osteoarthritis (OA) is a chronic degenerative disease in which slow and progressive damage to the articular cartilage, changes in the subchondral bone, formation of osteophytes, and alteration of the periarticular tissue result in inflammation and pain [1]. Although the causes of OA have not been completely identified, metabolic and inflammatory factors associated with age, gender, and genetic factors are known to contribute to OA by breaking down the articular cartilage [23]. Various factors responsible for the progression of OA, including proteolytic enzymes and inflammatory cytokines participating in the pathogenesis of OA, have been identified [456]. The inflammatory signaling pathway in OA is initiated by the cytokines IL-1β and TNF-α. Among the proinflammatory cytokines, both IL-1β and TNF-α are increased in the synovial fluid of patients with OA, and these two cytokines can directly or indirectly regulate and propagate inflammation. For instance, rabbits injected in the knee with IL-1 and TNF-α showed more substantive cartilage destruction than the control group injected alone [78910]. Nuclear transcription factor κB (NF-κB) or extracellular signal-regulated kinases (Erks) are involved in immune and inflammatory responses. They stimulate enzymes such as cyclooxygenase-2 (COX-2) and its subsequent products prostaglandin E2 (PGE2), nitric oxide (NO), and matrix metalloproteinases (MMPs), which ultimately results in the degeneration of cartilage tissue [11121314]. Moreover, the expression of MMP-3, MMP-7, and MMP-9 is enhanced at the onset of arthritis, and the increase in these MMPs exacerbates OA by damaging the collagen matrix composing the cartilage [151617].
OA results in pain, stiffness, and swelling, leading to difficulties with activities and motor abnormalities in daily living, eventually decreasing the quality of life [18]. Currently, there is no treatment available that can completely prevent OA. Current management strategies mainly include the use of paracetamol, analgesic drugs, and non-steroidal inflammatory drugs. Although these are the most effective modalities, various adverse effects, including acute drug toxicity, are to be considered. Another approach is intra-articular corticosteroid or hyaluronic acid injection, which minimizes and shortens the side effects [19]. Yet another way to manage OA involves physical therapy combined with a balanced diet or functional foods, as a lack in nutrients in the cartilage can result in joint degeneration or inflammation. One of the requirements for such functional foods is that they are to have an anti-inflammatory property or protective effect. According to recent studies, compounds such as omega-3 fatty acids, flavonoids, and carotenoids, which have anti-inflammatory effect, can alleviate or reduce arthritis progression [20]. However, there is still controversy about the effectiveness of these compounds; thus, further studies on the safety and efficacy of functional foods are required.
It is well known that H2O2 induces destruction of the extracellular matrix in chondrocytes and stimulate the expression of various inflammatory cytokines. In addition, reactive oxygen species (ROS) are produced by H2O2 and activate on the apoptosis of chondrocytes [21].
Among induced-OA animal models developed in previous studies, the animal model based on intra-articular injection of monosodium iodoacetate (MIA), in which the metabolism of chondrocytes is interrupted to damage the knee joint cartilage, showed symptoms similar to those of OA. The OA animal model, which also shows inflammatory responses in the synovium and the synovial fluid, has been recently widely used to assess the effectiveness of various chemicals for joint health [6]. In the present study, MIA was used to induce OA in rats.
Polydeoxyribonucleotide, a mixture of DNA fragments having a composition most similar to that of human DNA, is used for the treatment of scars and ulcers as well as postoperative tissue regeneration because it promotes cellular regeneration [222324]. It stimulates growth factors and extracellular matrix (ECM) turnover via purinergic adenosine A2A receptor in damaged cells or tissues. Previous studies on the effects of polydeoxyribonucleotide have shown that the activated A2A receptor prevents pro-inflammatory cytokines and promotes wound healing by releasing pro-fibrotic cytokines. The purpose of this study was to verify the anti-inflammatory effect of PRF001 in CHON-001 cells in which damage was induced by H2O2, and to assess the protective effect of PRF001 intake against arthritis in rats with MIA-induced arthritis.
Go to : 
METHODS
Chemicals
The polydeoxyribonucleotide, PRF001 (PRP salmon DNA; Pharma Research Products, Gangneung , Korea), used in the present study, is a fragmented DNA polymer extracted from the testes of adult chum salmons (Oncorhynchus keta, Salmonidae) that were returned to Namdae Stream, Yangyang, Gangwon, Republic of Korea over a period of three to five years through the salmon fry discharge project. PRF001, comprising molecular weights of 50 to 1,500 kDa, was dissolved at 0.9% to attain a DNA concentration above 75%. PRF001 was prepared at a specialized institute before use to satisfy all criteria with regard to lead (≤1 ppm), arsenic (≤1 ppm), cadmium (≤1 ppm), mercury (≤1 ppm), general bacteria (≤3,000 cells/g), and E. coli (negative). MIA and rotenone were obtained from Sigma (MO, USA).
Cell culture and in vitro study
The human chondrocyte cell line CHON-001 was obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were grown in Dulbecco's modified Eagle's medium:Ham's F12 (1:1 mixture) (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 µg/ml streptomycin (Gibco) in a 5% CO2 incubator at 37℃. To treat the cells, PRF001 was diluted in culture medium to the appropriate concentration.
Animal study
Male Sprague-Dawley rats (6–8 weeks old) purchased from Dae Han BioLink Co, Ltd, (Eumseong, Korea) were used in this study. The study protocol was reviewed and approved by the Animal Care and Use Committee of the Korea Institute of Science and Technology, Gangneung, Korea. The animals were acclimatized for approximately two weeks before the start of the experiments and were housed in normal stainless steel cages. The experimental rooms were maintained under standard conditions of temperature, relative humidity, ventilation, and illumination. Animals were fed ad libitum with a pelleted maintenance diet and allowed free access to drinking water. In all instances, the animals were handled humanely in accordance with the IACUC guidelines.
For induction of OA, rats were anesthetized using intraperitoneal anesthesia with mixture of Zoletile (cat. no. 06516; Virbac, Carros, France) and Rumpun (cat. no. 41882; Bayer Korea, Seoul, Korea). Then, the rats received an intra-articular injection of 50 µl MIA (60 mg/ml) into the right knee joint. For three days following the injection of MIA, the swelling and the walking abilities of the animals were monitored to verify the induction of arthritis. Rats were randomly divided into four groups of 10: a saline control group, a low-PRF001 concentration group (L) (10 mg/kg/day), a medium-PRF001 concentration group (M) (50 mg/kg/day), and a high-PRF001 concentration group (H) (100 mg/kg/day). Control rats received injections of 0.9% sterile saline. PRF001 was orally administered seven days after MIA injection. The rats in all groups were sacrificed after four weeks of oral PRF001 administration.
Cell viability assay
Cell viability was measured by a quantitative colorimetric assay using the EZ-Cytox Cell viability assay kit (Daeil Lab., Seoul, Korea). The assay provides a sensitive measurement of the metabolic status of the cells, particularly, of the mitochondria, as it is based on the cleavage of the tetrazolium salt to water-soluble formazan by the succinate-tetrazolium reductase system, which belongs to the respiratory chain of the mitochondria and is active only in viable cells. Briefly, exponentially growing cells were seeded in a 96-well plate at a density of 5×104 cells/well. The cells were pretreated with H2O2 for 2 h. Then, PRF001 was added to the culture medium at final concentrations of 0, 6.3, 12.5, 25, 50, and 100 µg/ml and the cells were incubated for 24 h. Control cells were left untreated. After the incubation, 10 µl of the EZ-Cytox assay kit reagent was added to each well, and the cells were incubated for 1 h. The absorbance at 450 nm in each well was measured with a microplate reader. The results are expressed as a percentage of the formazan dye absorbance of the control cells, which was set to 100%.
Measurement of intracellular reactive oxygen species (ROS)
Levels of intracellular ROS were estimated following treatment with the various compounds using 2′,7′-dichlorofluorescein diacetate (H2DCFDA) (Sigma, MO, USA) as a fluorescent probe. CHON-001 cells were exposed to PRF001 for 24 h, and the culture medium was replaced with fresh serum-free medium containing 20 µM DCFDA. The DCF fluorescence intensity was determined by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences, CA, USA) and CellQuest Pro software (BD Biosciences).
Biochemical tests of hepatocellular function
Blood samples were obtained from the inferior vena cava after four weeks of oral PRF001 administration. Serum alanine aminotransferase (sALT), serum aspartate aminotransferase (sAST), BUN, and creatinine levels were measured to assess hepatocyte injury using a Cobas C702 analyzer (Roche Diagnostics, Mannheim, Germany), per the manufacturer's instructions.
Histological analysis
For histopathological examination, cartilage tissues were collected, cut coronally, fixed in 10% formaldehyde, and embedded in paraffin. Five-micrometer sections were prepared and stained with hematoxylin and eosin (H&E).
Western blot analysis
The cartilages of rats were crushed in ice-cold lysis buffer (1 M Tris-HCl, pH 7.5, with protease inhibitors; 25 mM NaF; 10 mM NaV; 0.5 M EDTA; and 1% Triton X-100) and centrifuged at 14,000 rpm for 20 min. The protein concentration was determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA). Aliquots of 200 µg of protein extract were separated by 10%–15% sodium dodecyl sulfate polyacrylamide gel electrophorese (SDS-PAGE; Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% milk in Tris-buffered saline and Tween-20 buffer (10 mM Tris, 100 mM NaCl, and 0.1% Tween-20, pH 8.0) and subsequently probed with primary antibodies against heat-shock protein 70 (HSP-70), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), IL-1β, NF-κB, TNF-α, MMP3/7/9 (1:1,000; Abcam, Cambridge, MA, USA), inducible nitric oxide synthase (iNOS, 1:1,000 dilution, BD Transduction Laboratory, San Diego, CA, USA), p-Erk1/2, COX-2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4℃. Membranes were then probed with goat anti-rabbit (1:1,000) or goat anti-mouse (1:1,000) horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). The protein bands were detected using the EZ-Capture ST imaging system (Atto, Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of PGE2 and TNF-α were measured using specific ELISA kits (BD Biosciences). Briefly, 2×105 cells/well in 12-well plates were treated or not with H2O2 and with PRF001 for 24 h, the supernatants were collected, and PGE2 and TNF-α were quantified according to the manufacturer's protocol, using standard curves.
Statistical analysis
All data are presented as the mean±standard error of mean (SEM) and were evaluated by one-way analysis of variance (ANOVA) with Bonferroni's post-hoc correction (SPSS software, version 15.0; SPSS Inc., Chicago, IL, USA). p-values <0.05 were considered significant.
Go to :
RESULTS
PRF001 protects CHON-001 cells against damage induced by H2O2
To determine the toxicity of PRF001 on CHON-001 human chondrocyte cells, cells were exposed to PRF001 at different concentrations (6.3, 12.5, 25, 50, and 100 µg/ml) and their viability was measured by MTT assay. As shown in Fig. 1A, the viability of PRF001-treated cells at all concentrations tested was maintained at the level observed in the control cells, indicating that PRF001 has no toxic effect on the cells.
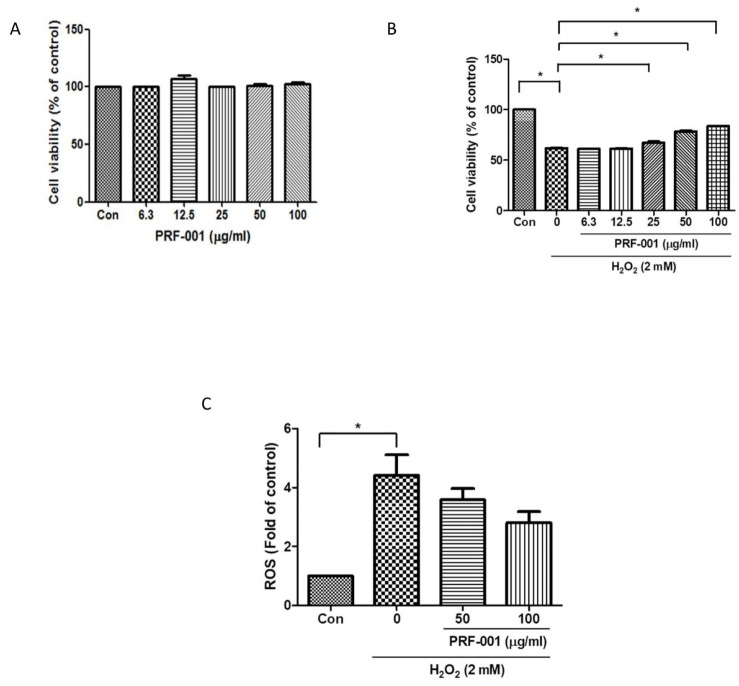 | Fig. 1PRF001 is not toxic to CHON-001 cells and improves the viability of and H2O2-induced CHON-001 cells in a concentration-dependent manner.(A) Cytotoxicity of PRF001 on CHON-001 cells. PRF001 was added to CHON-001 cells at 6.3, 12.5, 25, 50, or 100 µg/ml for 24 h. Cell viability was measured in supernatants of treated CHON-001 cells and expressed as a percentage of untreated control cells in each group. (B) Cell viability in H2O2-induced CHON-001 cells. H2O2 was added to CHON-001 cells for 2 h followed by treatment with PRF001 for 24 h. Cell viability was measured in supernatants of treated CHON-001 cells and expressed as a percentage of untreated control cells in each group. (C) ROS level in H2O2-induced CHON-001 cells. H2DCFDA was added to CHON-001 cells after treatment with PRF001 for 24 h. ROS levels were measured as DCF fluorescence intensity in supernatants of 50 and 100 µg/ml-treated CHON-001 cells by flow cytometry. The data are expressed as the mean±SEM of triplicate experiments. *Statistically significant, p<0.05.
|
Next, the protective effect of PRF001 against H2O2-induced damage in CHON-001 was examined. Cells were treated with H2O2 and then incubated with PRF001 at the same concentrations as those used for the cytotoxicity assay. As illustrated in Fig. 1B, the viability of CHON-001 cells was decreased approximately 30% by treatment with H2O2. There was no increase in cell survival at low concentrations (6.3 and 12.5 µg/ml) of PRF001; however, the viability of CHON-001 cells significantly increased dose-dependent at concentrations of PRF001 ranging from 25 to 100 µg/ml.
ROS and oxidative stress are known to induce cell apoptosis. Therefore, the level of ROS induced by H2O2 as an inflammatory response was analyzed in CHON-001 cells. Cells were pretreated with PRF001 at concentrations of 50 and 100 µg/ml for 24 h and then incubated with DCFDA for 1 h. After the cells were treated with H2O2 for 30 min, intracellular ROS levels were measured using FACS analysis. The ROS level was elevated by nearly 4.5-fold in CHON-001 cells induced by H2O2, as compared to the control cells (Fig. 1C). However, cells pretreated with 50 and 100 µg/ml PRF001 exhibited a decrease in ROS production though not significantly difference.
Taken together, these results demonstrated that PRF001 shows no cytotoxicity at concentrations up to 100 µg/ml and it protects cells against H2O2-induced damage (Fig. 1B).
PRF001 reduces inflammatory mediators and cytokines in H2O2-induced CHON-001 cells
To investigate whether the protective effect in CHON-001 cells was associated with inflammatory mediators and cytokines, the protein levels of IL-1β and TNF-α, a key inflammatory marker, were measured by western blot analysis. Cells were exposed to H2O2 and PRF001 at concentrations of 50 and 100 µg/ml for 24 h. IL-1β expression was enhanced by approximately 4-fold in cells exposed to H2O2 as compared to control cells ; yet this expression was not considerably decreased by PRF001 (50 and 100 µg/ml). However, the expression of TNF-α significantly decreased in cells treated with 50 and 100 µg/ml PRF001 (Figs. 2A and G) which was induced by H2O2.
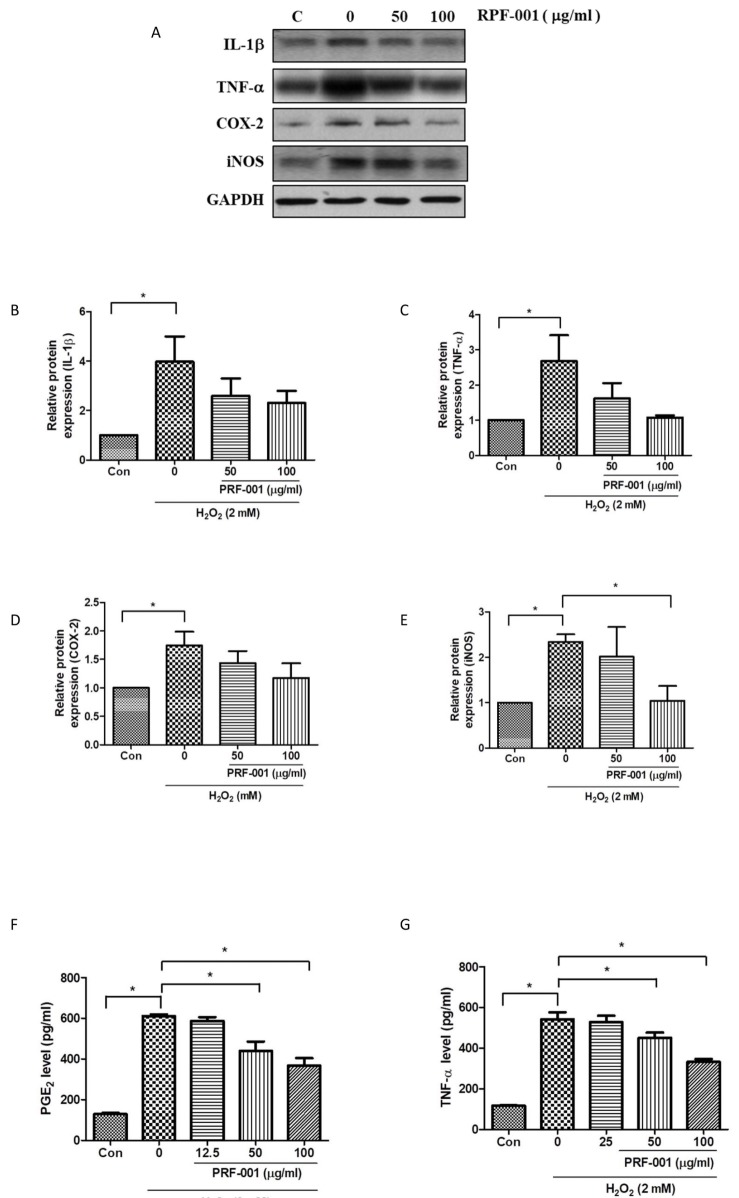 | Fig. 2PRF001 lowers inflammatory mediators and cytokines in H2O2-induced CHON-001 cells.(A–E) CHON-001 cells were treated with H2O2 and PRF001 at concentrations of 50 and 100 µg/ml for 24 h. Protein levels of IL-1β, TNF-α, COX-2, and iNOS were measured in cell lysates using western blot assay. (F, G) CHON-001 cells were pretreated with H2O2 for 2 h, followed by treatment with PRF001 at concentrations of 50 and 100 µg/ml for 24 h. Protein levels of PGE2 and TNF-α were measured in cell supernatants by ELISA. The data are expressed as the mean±SEM from triplicate experiments. *Statistically significant, p<0.05.
|
Elevated COX-2 and NO have been observed in synovial fluid of patients with OA. Thus, the protein levels of COX-2 and iNOS in CHON-001 cells were analyzed by western blot assay. As shown in Figs. 2D and E, both COX-2 and iNOS protein levels were markedly increased in cells exposed to H2O2 alone as compared to control cells. The expression of COX-2 was slightly reduced in cells treated with 100 µg/ml. However, iNOS expression was decreased in PRF001-treated cells, particularly, a marked decrease in 100 µg/ml PRF001-treated cells was observed.
PGE2, an OA accelerator, is stimulated by inflammatory mediators such as IL-1β, TNF-α, and COX-2, and PGE2 released from articular chondrocytes induces cartilage degradation in human patients with OA. To examine whether the protective mechanism of PRF001 involves modulation of inflammatory mediators and cytokines, PGE2 and TNF-α protein levels were measured by ELISA. CHON-001 cells were induced with 2 mM H2O2 for 2 h and then exposed to PRF001 for 24 h. As shown in Fig. 2F, the level of PGE2 was greatly increased in cells exposed to H2O2 alone, while it was significantly decreased after treatment with 50 µg/ml PRF001, and even more so after treatment with 100 µg/ml PRF001. These results demonstrated that PRF001 has anti-inflammatory properties against H2O2 induction in vitro.
PRF001 decreases the production of inflammatory mediators and cytokines in a MIA-induced OA rat model
IL-1β is a cytokine involved primarily in the onset of OA. To investigate whether the IL-1β level is affected by PRF001 in MIA-induced OA rats, rats were orally administered low (L), medium (M), or high (H) doses of PFR001 for four weeks, and cartilage tissues were harvested from all groups for western blot assay. As shown in Fig. 3B, the protein expression of IL-1β in the MIA-induced group was increased 2.5-fold as compared to the control group, however IL-1β expression was considerably decreased in both MIA+M and MIA+H group. P-Erk1/2 and NF-κB are involved in the signaling pathway related to inflammatory cytokine activation in the process of OA. Both p-Erk1/2 and NF-κB were induced by MIA compared to the control group. There was a significant decrease in the expression of p-Erk1/2 in all group of MIA+L, MIA+M and MIA+H, while dose-dependently decreased in the expression of NF-κB (Figs. 3C and D).
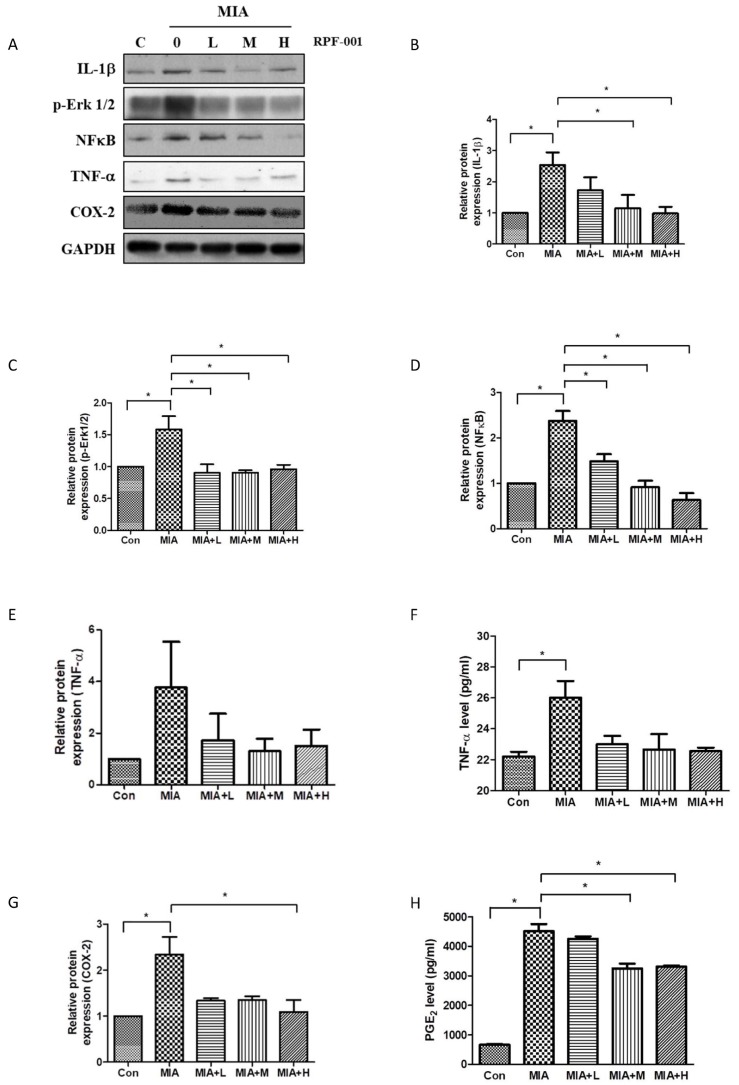 | Fig. 3PRF001 diminishes the production of inflammatory mediators and cytokines in MIA-induced OA rats.(A–E, G) MIA (60 mg/ml) was injected into the right knee joint of rats, and seven days later, PRF001 was administered orally at different concentrations (low, medium, and high; 10, 50, and 100 mg/kg/day) for four weeks. Protein levels of IL-1β, Erk, NF-κB, TNF-α, and COX-2 were measured in the cartilage tissue of all groups using western blot assay. (F, H) Protein levels of TNF-α and PGE2 were measured in sera of all groups using ELISA. The data are expressed as the mean±SEM of ten animals per group. *Statistically significant, p<0.05.
|
To analyze whether the expression of these mediators was involved in the production of inflammatory cytokines, TNF-α protein levels in cartilage tissues of all groups were measured. TNF-α protein level in serum was significantly increased in the group treated with MIA, while it was slightly decreased in MIA+M and MIA+H groups (Figs. 3E and F).
The enzyme COX-2 is activated by inflammatory cytokines at the inflammation site during OA. As a result of MIA-induction, COX-2 protein was overexpressed; however, the expression was decreased in MIA+L, MIA+M and MIA+H, with the most marked decrease observed in MIA+H group. Considering the fact that COX-2 modulates PGE2 levels during inflammation, PGE2 levels were measured by ELISA. An elevated PGE2 level was detected in the MIA-induced group, which was significantly decreased in both MIA+M and MIA+H. These results showed that PRF001 has an anti-inflammatory effect by decreasing the production of inflammatory mediators and cytokines in MIA-induced OA rats (Figs. 3G and H).
PRF001 regulates catabolic activity by reducing MMP expression in MIA-induced OA rats
The expression of MMP3 and MMP7, which are involved in catabolic activity of chondrocytes, in OA model rats treated with the various concentrations of PRF001 was assessed by western blot assay. As shown in Fig. 4, the protein expression of both MMP3 and MMP7 was approximately 2-fold higher in the MIA-induced than in control animals, which means that chondrocytic catabolic activity was upregulated by MIA. However, the increase in MMP3 expression was greatly decreased both in MIA+M and MIA+H groups. Also, MMP7 expression was significantly decreased in MIA+L as well as MIA+H group (Figs. 4B and C).
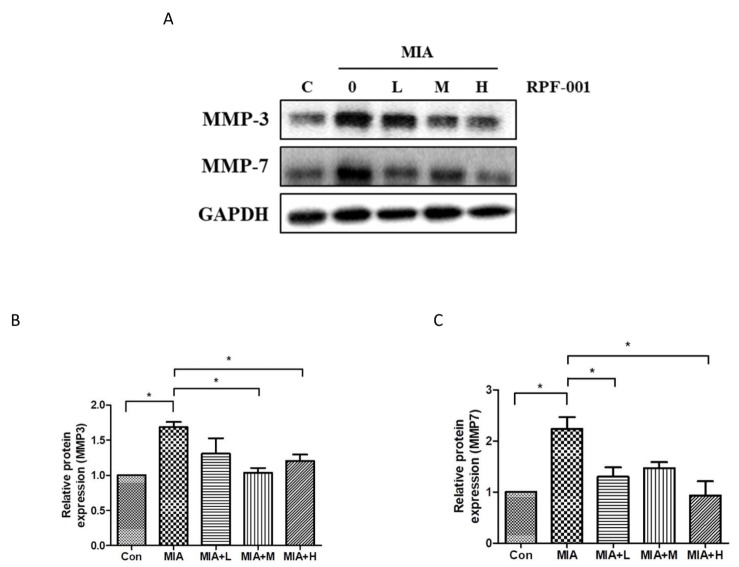 | Fig. 4PRF001 is involved in catabolic activity of chondrocytes by diminishing the MMP expression in MIA-induced OA rats.MIA (60 mg/ml) was injected into the right knee joint of rats, and seven days later, PRF001 was administered orally at different concentrations (low, medium, and high; 10, 50, and 100 mg/kg/day) for four weeks. Protein levels of MMP3 and MMP7 were measured in the cartilage tissue of all groups using western blot assay (A, C). The data are expressed as the mean±SEM for ten animals per group. *Statistically significant, p<0.05.
|
PRF001 protects articular cartilage and subchondral bone in MIA-induced OA rats
To validate the effect of PRF001 on OA knee joints, cartilage tissues were harvested from animals of all treatment groups and stained with H&E for histological analysis. As shown in Fig. 5, chondrocytes were uniformly distributed in parallel rows in the transition zone of articular cartilage in the control group, whereas cartilage degeneration with irregular distribution as well as changes in chondrocyte morphology was revealed in the MIA-induced rats. On the other hand, treatment of medium-concentration of PRF001 restored the irregularity in chondrocyte distribution, while high-concentration of PRF001 greatly improved the distribution as well as chondrocyte morphology.
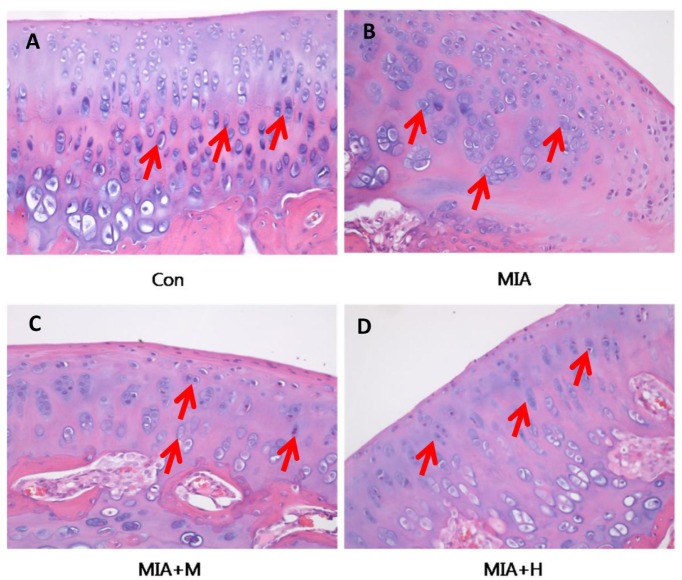 | Fig. 5PRF001 protects articular cartilage and subchondral bone in MIA-induced OA rats.Coronal sections of cartilage tissues were stained with H&E. (A) Evenly distributed chondrocytes were observed in the normal control group. (B) Changes in morphology and distribution of chondrocytes in the MIA group and (C, D) concentration-dependent protective effects revealed in PRF001-administered groups (MIA+M, H). The data are expressed as the mean±SEM of 10 animals per group.
|
Go to :
DISCUSSION
OA, joint disease, or degenerative arthritis, is a common chronic condition of the joints caused by internal or external stress, such as inflammatory chemicals (cytokines), proteolytic enzymes, and repeated overuse of the joints. While several preand clinical studies have assessed the significance of targeting cellular mechanisms, studies of polydeoxyribonucleotide in OA animal models are lacking. Therefore, the specific purposes of the present study were to examine i) the potential anti-inflammatory property of PRF001 in H2O2-damaged chondrocytes in vitro, ii) the protective effect of oral administration of PRF001 in OA rats.
MIA induces local inflammation from the synovial fluid to the membrane of the knee joint, which results in the production of inflammatory mediators such as IL-1β and TNF-α. These mediators further promote the pathogenesis of OA by increasing inflammatory cytokines and MMPs, which is followed by chondrocyte apoptosis and cartilage degradation [252627]. In this research, MIA-induced OA in rats was confirmed by increases in inflammatory mediators and cytokines, such as IL-1β, p-Erk1/2, NF-κB, and TNF-α, which play crucial roles in the process of OA. Our results revealed that oral administration of PRF001 in the MIA-induced OA rats induced declines in the above markers. Furthermore, the expression of MMP3 and MMP7, which are involved in catabolic activity of chondrocytes leading to the degradation of cartilage components, including collagen and proteins, were also decreased. Considering these findings, a high dose of PRF001 has protective effects in H2O2-induced chondrocytes as well as in MIA-induced OA rats.
Polydeoxyribonucleotide , a major component of PRF001, is a fragmented DNA polymer known as a tissue-stimulating agent. Polydeoxyribonucleotide has been proven to regulate the levels of cytokines such as TNF-α, IL-2, and IL-6 via the A2A receptor signaling cascade both in vitro and in vivo [2829]. Consistent with these studies, we found that inflammatory mediators and cytokines were regulated by PRF001 in both H2O2-induced chondrocytes and MIA-induced OA rats. In addition, treatment with PRF001 was verified to prevent the activation of proteolytic enzymes such as MMPs.
IL-1β activation is mediated by the membrane receptors IL-R1 and IL-R2 [4]. Once IL-1β is activated by binding to IL-R1, it activates transforming growth factor beta-activated kinase 1 (TAK1), which in turn activates transcription factor MAPK and NF-κB by regulating I κB kinase complex (IKK) [5]. Subsequently, iNOS is stimulated to produce NO, resulting in the apoptosis of chondrocytes. TNF-α and IL-1β inhibit the synthesis of proteoglycans and collagens, which are the major components of ECM in articular cartilage, and stimulate MMP activity, resulting in accelerated degradation of cartilage tissues [730]. Interestingly, our results suggested that PRF001 suppresses the expression of IL-1β and TNF-α as well as p-Erk1/2 and NF-κB, subsequently reducing iNOS expression.
Inflammatory mediators such as IL-1β and TNF-α stimulate signal molecules downstream of the OA pathway. In particular, COX-2 expression is upregulated in the chondrocytes, resulting in increased levels of PGE2, which subsequently stimulate the expression of MMPs in damaged chondrocytes [31323334]. Accordingly, our study showed the increased expression of COX-2 and PGE2 in H2O2 and MIA-induced OA models, whereas PRF001 suppressed these responses. Activated p-Erk1/2, NF-κB and inflammatory factors such as COX-2 and PGE2 further function in the progress of OA by inducing MMPs [35]. Activated MMP3, known as stromelysin, stimulates A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS5), which are critical to the deterioration of proteoglycans, causing severe cartilage matrix damage [36]. Likewise, high MMP7 expression has been shown to be involved in catabolic activity in progressive OA [37]. In agreement with these studies, our data demonstrated that both MMP3 and MMP7 were significantly upregulated in MIA-induced OA rats; however, PRF001 effectively reduced this response.
In the OA pathologic process, the inflammatory mediators and MMPs induce changes in the morphology of chondrocytes, considered as an ultimate manifestation of intracellular processes in OA. In support of this, MIA-induced OA rats showed changes in the morphology and distribution of chondrocytes, resulting in severe damage in articular cartilage. However, OA rats treated with a high dose of PRF001 exhibited normal chondrocyte morphology and distribution.
This study had several limitations. First, specific articular structures and the presence of osteoclasts, the obvious indicator of OA, as well as fragmentation of subchondral bone should be analyzed via micro-CT in future. Second, the suppressive effect of PRF001 on pain, one of the important manifestations in the progress of OA, should be determined [38]. Lastly, further studies are required to elucidate the mechanism of PRF001 under arthritic conditions, and whether or not it is related to adenosine receptors.
In conclusion, to our knowledge, this study is the first to show the anti-inflammatory property of PRF001, and that this contributes to its protective effects in OA through deregulating IL-1β, TNF-α and subsequent signals, including p-Erk1/2, NF-κB, COX-2, PGE2, and MMPs. Our findings indicate that this polydeoxyribonucleotide has potential for therapeutic application.
Go to :
 XML Download
XML Download