

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Depression can be a devastating illness that results in personal disability and has been associated with altered brain structure and function [1]. Depression can be caused by many factors and results in structural, functional, and molecular changes in many regions of the brain including the prefrontal cortex (PFC), hippocampus, nucleus accumbens, and amygdala [234]. Chronic stress leads to neuronal atrophy and reduced synaptic functioning in animal models [56]. It has also been shown to downregulate hippocampal brain-derived neurotrophic factor (BDNF) expression [7].
On the molecular level, depression results from epigenetic consequences of environmental and genetic factors. HDACs are enzymes that remove acetyl groups from lysines on histone tails. Of the four class IIa HDACs (HDAC4, 5, 7, and 9), HDAC5 activity has been shown to be increased by chronic stress in the rodent hippocampus and in the human postmortem hippocampus; HDAC5 activity was returned to normal levels by antidepressant administration [8]. Phosphorylation of HDAC5 by HDAC5 kinases induces nuclear export of phosphorylated HDAC5, thereby activating gene expression [910]. Ketamine, a non-competitive NMDA receptor antagonist, has been shown to produce a rapid antidepressant-like effect through phosphorylation-dependent nuclear export of HDAC5 in the rat hippocampus [11].
3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP), a rigid analog of 2-amino-7-phosphonoheptanoate (AP7), is a competitive NMDA receptor antagonist [12]. CPP has been shown to exert antidepressant-like behavioral effects in the forced swim test (FST) in an animal model of depression [13]. In addition, low-dose administration of CPP was shown to inhibit stress-induced dendritic retraction in the medial prefrontal cortex (mPFC) [14]. However, the mechanism by which CPP induces these antidepressant-like effects is unclear. In the present study, we found that CPP increases HDAC5 phosphorylation and attenuates HDAC5-mediated repression of Bdnf transcription in the hippocampus. We further show that HDAC5 knockdown in the hippocampus prevents CPP-mediated behavioral actions in stressed rats, indicating that CPP-mediated antidepressant responses include HDAC5 pathways.
Go to : 
METHODS
Animals
Adult male Sprague-Dawley (SD) rats (8–10 weeks old; Charles River Laboratories, Wilmington, MA, USA) weighing 250 to 280 g were pair-housed and maintained on a 12-h light-dark cycle with access to food and water ad libitum. All procedures were in strict accordance with Institutional Animal Care and Use Committee (IACUC) guidelines, and the use of laboratory animals was approved by the Hanyang University Animal Care and Use Committee. All animals were randomly assigned to one of the experimental groups.
Drug treatment
3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP, Sigma) was dissolved in 0.9% saline at a concentration of 0.5 mg/kg and was injected intraperitoneally. In the controls, 0.9% saline was injected intraperitoneally.
Chronic unpredictable stress (CUS) procedure
To simulate CUS, animals were subjected to the exact sequence of 12 stressors (2 per day for 28 days) described in Koo et al. [15]: light on, light off, cage tilt, cage rotation, wet bedding, restraint, strobe, odor, food deprivation, overcrowding, cold (4℃), and isolation.
Forced swim test (FST)
The FST is one of the most common behavioral experiments used to assess depression-like behavior in rodents. In this test, rats are forced to swim for 10 min in a cylinder from which there is no escape, after which the immobility time is measured. Immobility (passive) behavior was defined as floating without any motion, while swimming across the cylinder or climbing the wall of the cylinder was considered active behavior. All FSTs were recorded with a camcorder for subsequent video analysis.
Locomotor activity test (LMA)
The locomotor activity of rats was evaluated by an automatic video tracking system (SmarTrack®, Smartech, Madison, WI). Briefly, rats were placed in the marginal part of the open-field arena (76.5 cm×76.5 cm×40 cm) and allowed to explore the arena for 10 min. The total distance travelled in 10 min was measured by the video tracking system.
Novelty suppressed feeding test (NSFT)
Animals were food deprived for 12 h and on the test day were placed in an open field (76.5 cm×76.5 cm×40 cm) with eight pellets of food in the center. The animals were given 8 min to approach the food and eat. The test was stopped as soon as the animal took the first bite. The latency to eat was recorded in seconds. Home cage food intake was also measured as a control.
Chromatin immunoprecipitation (ChIP) assay
Rat hippocampal tissues were fixed with 1% formaldehyde in PBS. Lysates were sonicated and extracted DNA was cut to lengths between 200-500 bps. The sonicated samples were rotated to preclear with protein A agarose (Roche Applied Sciences) for 1 h at 4℃ and anti-HDAC5 antibody (Abcam) was added and incubated overnight at 4℃ followed by incubation with fresh Protein A agarose for 2 h at 4℃. Precipitated chromatin complexes were eluted from the beads. Finally, the protein-DNA cross-links were incubated with protease K (Roche Applied Sciences) for 2 h at 42℃ and immunoprecipitated DNA was purified. Purified DNA samples were normalized and subjected to PCR analysis.
Quantitative real-time PCR
Immunoprecipitated DNA samples were resuspended in H2O and fractions used for real-time PCR (iCycler, Bio-Rad, Hercules, CA, USA). Input (non-immunoprecipitated DNA) and immunoprecipitated DNA were PCR amplified in triplicate in the presence of SYBR Green (Bioline, London, UK). Ct values for each sample were obtained using Sequence Detector 1.1 software. QPCR data were analyzed by ΔΔCt method.
Western blot analysis
Equal amounts of protein extracts (20–25 µg) were mixed with SDS sample buffer and denatured at 95℃. The proteins were then electrophoresed on 7–12% polyacrylamide gels and transferred onto nitrocellulose membrane filters (Amersham Pharmacia Biotech, Amersham, UK). Blots were blocked with 1-5% non-fat milk in 1X TTBS (0.1% Tween-20 in 1X TBS) for 1 h at room temperature and incubated overnight at 4℃ with primary antibodies.
Virus production and purification
Virus was generated using a triple-transfection, helper-free method and purified according to a published protocol (Invitrogen). Briefly, HEK 293T cells (ATCC, Manassas, VA, USA) were cultured in five 150×25 mm cell culture dishes and transfected with the ViraPower lentiviral expression system (Invitrogen) using Lipofectamine (Invitrogen). Cells were collected, pelleted, and resuspended in buffer (0.15 M NaCl and 50 mM Tris, pH 8.0) at 66–70 h after transfection. After two freeze-thaw cycles to lyse the cells, benzonase was added (final concentration, 50 U/ml), and the mixture was incubated at 37℃ for 30 min. The lysate was added to PEG-it solution and processed according to the manufacturer's protocol (System Biosciences, Inc., Mountain View, CA, USA). The final purified virus was stored at −80℃.
Stereotaxic surgery and infusions
Rats were anesthetized with xylazine hydrochloride (25 mg/kg) and tiletamine hydrochloride/zolazepam hydrochloride (50 mg/kg). Purified virus was injected bilaterally into the dentate gyrus (DG) at coordinates −4.1 mm (anterior/posterior), ±2.4 mm (lateral), and −4.6 mm (dorsal/ventral) relative to bregma. A total of 6 µl of purified virus was delivered at a rate of 0.1 µl/min, followed by 10 min of rest. The needle was removed, and the scalp incision was closed with wound clips. Rats in the same cage were randomly assigned to different experimental groups for the behavioral study; the order of testing was distributed across groups.
Immunohistochemistry
After animals were perfused with PBS, their brains were fixed overnight in 4% paraformaldehyde and then transferred to 30% sucrose. Brain sections (30 µm thickness) were cut using a microtome for visualization of EGFP. Gene transfer efficiency was assessed by immunofluorescence staining 4 weeks after surgery. EGFP staining was observed predominantly in dentate granule cells surrounding the infusion site in each hemisphere. Anti-GFP monoclonal antibodies were used (1:300, Roche). Slices were incubated in secondary antibodies conjugated to Alexa Fluor 488 (1:300, Invitrogen) and then mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) to preserve fluorescence. Images were captured using a fluorescence microscope (Nikon, Tokyo, Japan).
Statistical analysis
All data were obtained from at least three independent experiments. Data are presented as mean±S.E.M. Statistical significance was calculated by unpaired Student's t-test. *p<0.05, **p<0.01, ***p<0.001. For the behavioral experiment, the statistical significance of differences between the four groups was determined by analysis of variance (ANOVA; StatView 5, SAS Software, Cary, NC, USA) followed by LSD post hoc analysis.
Go to :
RESULTS
CPP rapidly induced HDAC5 phosphorylation in a time-dependent manner and triggered cytoplasmic localization of HDAC5 in the rat hippocampus
We first investigated whether CPP induces HDAC5 phosphorylation and determined when CPP-mediated HDAC5 phosphorylation reached its peak level. To this end, 8-week-old rats were treated intraperitoneally for various lengths of time with 0.5 mg/kg CPP. This concentration was chosen because antidepressant-like behaviors were previously observed in the FST at 0.5 mg/kg [13]. Since HDAC5 phosphorylation at Ser259 and Ser498 has been shown to result in nuclear export of HDAC5 by 14-3-3 proteins [16], the levels of HDAC5 phosphorylation at Ser259 and at Ser498 were measured by immunoblotting. Intraperitoneal administration of CPP to the rats induced phosphorylation of HDAC5 at Ser259 and Ser498 in a time-dependent manner, with peak phosphorylation at approximately 0.5 h (Figs. 1A and B).
 | Fig. 1CPP rapidly induced HDAC5 phosphorylation in a time-dependent manner and triggered cytoplasmic localization of HDAC5 in the rat hippocampus.(A) CPP (0.5 mg/kg) increased phosphorylation of HDAC5 in the rat hippocampus in a time-dependent manner. Peak phosphorylation was observed at approximately 0.5 h after treatment. (B) Quantitative analysis of the HDAC5 phosphorylation shown in (A) (n=3 per group). (C) CPP (0.5 mg/kg) induced phosphorylation of cytoplasmic HDAC5 at 1 h post-treatment. (D) Quantitative analysis of the HDAC5 phosphorylation in cytoplasm shown in (C) (n=7 per group). Values are expressed as mean±S.E.M. Student's t-test, *p<0.05, **p<0.01, ***p<0.001 compared to the CTL group.
|
We next evaluated whether cytoplasmic localization of HDAC5 is triggered by CPP at 1 h. Rat hippocampal tissue extracts were separated into cytoplasmic and nuclear fractions, and the levels of HDAC5 phosphorylation at Ser259 and Ser498 were measured in the cytoplasmic fractions. The results showed that CPP rapidly increased phosphorylation of HDAC5 at Ser259 and Ser498 in the rat hippocampal cytoplasm by 1 h post-treatment (Figs. 1C and D). Therefore, these results indicate that CPP rapidly induces HDAC5 phosphorylation, and that the phosphorylated HDAC5 translocates to the cytoplasm. These observations suggest that phosphorylated HDAC5 initiates the transcription of genes associated with antidepressant-like effects.
CPP increased phosphorylation of PKD and CaMKII and upregulated BDNF expression
Phosphorylation of HDAC5 has been reported to be regulated by CaMKII activity in neurons [1718] and by protein kinase D (PKD) in other cell types [1920]. To determine whether HDAC5 phosphorylation in response to CPP is associated with PKD and CaMKII in the rat hippocampus, the levels of phosphorylated PKD and CaMKII induced by CPP were investigated by immunoblotting. Western blotting revealed that CPP stimulated phosphorylation of PKD and CaMKII in hippocampus at 1 h post-treatment (Figs. 2A and B). The phosphorylation of PKD and CaMKII were increased in a time-dependent manner with a peak level at approximately 10-30 min after injection, a time point that is earlier than that of HDAC5 phosphorylation (Figs. 2C and D). These indicate that phosphorylation of PKD and CaMKII was increased before the induction of HDAC5 phosphorylation and suggest that CPP-mediated phosphorylation of HDAC5 might be regulated by a PKD- and CaMKII-dependent pathway.
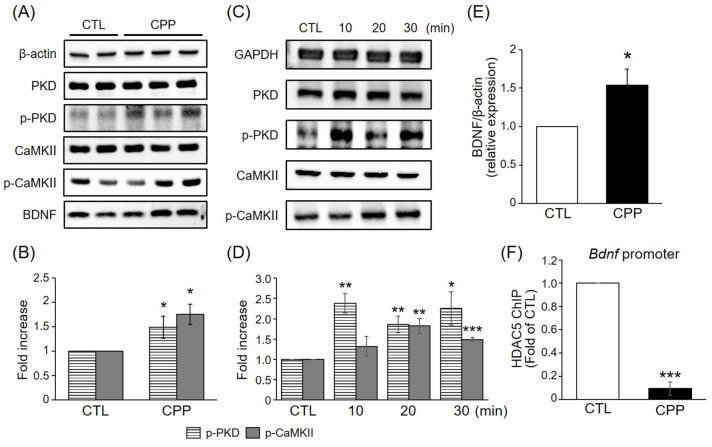 | Fig. 2CPP increased phosphorylation of PKD and CaMKII and upregulated BDNF expression.(A) Representative immunoblots of hippocampal extracts of rats exposed to CPP (0.5 mg/kg). Extracts were generated 1 h after treatment. Protein levels were normalized to that of β-actin. (B) Quantitative analysis of p-PKD and p-CaMKII levels. Values were normalized to that of total PKD or CaMKII and expressed relative to CTL (n=6 per group). (C) CPP (0.5 mg/kg) increased phosphorylation of PKD and CaMKII in the rat hippocampus in a time-dependent manner. Peak phosphorylation of PKD and CaMKII was observed at approximately 10 min and 20 min respectively after treatment. (D) Quantitative analysis of the PKD and CaMKII phosphorylation is shown in (C) (n=3 per group). (E) Quantitative analysis of BDNF levels shown in (A). Values were normalized to that of β-actin (n=6 per group). (F) ChIP assays. Decreased binding of HDAC5 to the Bdnf promoter in the hippocampus of rats treated with CPP (0.5 mg/kg) (n=3 per group). Values are expressed as mean±S.E.M. Student's t-test, *p<0.05, **p<0.01, ***p<0.001 compared to the CTL group.
|
BDNF plays a key role in neuroprotective and neurotrophic processes, which are relevant to stress and mood disorders [2122]. CPP rapidly increased the protein level of BDNF at 1 h (Figs. 2A and E). To determine whether CPP regulates BDNF gene expression through HDAC5 in hippocampal neurons, the DNA binding activity of HDAC5 to the Bdnf gene promoter was measured by ChIPs in hippocampus. HDAC5 was less enriched at the Bdnf promoter in hippocampal neurons treated with CPP than in those cells treated with saline (Fig. 2F). Given that association with HDAC5 in the specific promoter regions is linked to their repression, these results were consistent with the induction of BDNF proteins by CPP. These results suggest that CPP increases HDAC5 phosphorylation through a PKD- and CaMKII-dependent pathway, thereby inducing neuroprotective and neurotrophic processes that ameliorate depression-like behavior and CPP modulates BDNF transcriptional activity through HDAC5.
CPP had a rapid antidepressant-like effect in the FST and the NSFT
Exposure of rats to CUS has consistently been shown to induce cognitive impairment and to reduce the rate of body weight gain over the course of CUS exposure. These effects are similar to the components of major depression and anxiety disorders [23]. To investigate whether CPP has antidepressant-like effects on behavior, 8-week-old rats were exposed to a 4-week CUS regimen or a control period. The FST and NSFT were then conducted 1 h after intraperitoneal CPP injection (0.5 mg/kg) (Fig. 3A).
 | Fig. 3CPP had a rapid antidepressant-like effect in the FST and the NSFT.(A) Schematic diagram of the experimental design. Rats were subjected to chronic unpredictable stress (CUS) for 28 days. CPP (0.5 mg/kg) was administered 1 h before the FST and NSFT. (B) The mean body weight of stressed rats was lower than that of nonstressed rats (Nonstressed, n=17; CUS, n=15). Values are expressed as mean±S.E.M. Student's t-test, ***p<0.001 compared to the CTL group. (C) Immobility times in the nonstressed and CUS groups at the early stage of the FST (5 min). Main effect of CPP: F1,28=13.637, p<0.01; main effect of stress: F1,28=0.024, p>0.05; interaction: F1,28=0.56, p>0.05. (D) Immobility times in the nonstressed and CUS groups at the late stage of the FST (10 min). Main effect of CPP: F1,28=6.939, p<0.05; main effect of stress: F1,28=0.526, p>0.05; interaction: F1,28=3.218, p>0.05 (Nonstressed CTL, n=9; Nonstressed CPP, n=8; CUS CTL, n=7; CUS CPP, n=8). (E) NSFT. Main effect of CPP: F1,16=4.476, p=0.05; main effect of stress: F1,16=1.119, p>0.05; interaction: F1,16=3.841, p>0.05 (Nonstressed CTL, n=5; Nonstressed CPP, n=5; CUS CTL, n=5; CUS CPP, n=5). (F) Home cage feeding test. Main effect of CPP: F1,16=0.008, p>0.05; main effect of stress: F1,16=0.006, p>0.05; interaction: F1,16=0.239, p>0.05 (n=5 per group). (G) Locomotor activity. Main effect of CPP: F1,16=0.393, p>0.05; main effect of stress: F1,16=3.038, p>0.05; interaction: F1,16=0.132, p>0.05 (n=5 per group). Values are expressed as mean±S.E.M. Two-way ANOVA was followed by LSD post hoc analysis. *p<0.05, ***p<0.001 compared to nonstressed control rats. #p<0.05, ##p<0.01 compared to CUS control rats.
|
Consistent with a previous study [23], the mean body weight of the CUS group was significantly lower than that of the non-stressed group (Fig. 3B), indicating that the CUS-exposed rats were affected by the various stressors. In the FST, which uses decreased immobility time as an index of antidepressant efficacy, the immobility time was reduced by CPP in the nonstressed group and in the CUS group in the early stage (5 min) (Fig. 3C). However, at the late stage of the FST (10 min), the immobility time was only decreased by CPP in the CUS group (Fig. 3D). In the NSFT, which uses the latency period to feeding as an index of anxiety and antidepressant-like response, CPP reduced latency to feeding in the CUS group (Fig. 3E), consistent with a previous report on anxiolytic behavior in animals injected with ketamine [11]. There was no difference in the home cage food intake between groups (Fig. 3F). In the locomotor activity test (LMA), which uses total distance moved as an index of motor function, there was no difference between groups (Fig. 3G). When combined with the results demonstrating an effect of CPP on antidepressant-like behaviors in the FST, these behavioral results indicate that CPP rapidly induces an antidepressant response that is more effective under stress conditions.
HDAC5 knockdown produced antidepressant effects and occluded the actions of CPP in chronically stressed rats
To further address the role of HDAC5 in the behavioral response to chronic stress and CPP, a lentivirus expressing shRNA targeted against rat HDAC5 (lenti-shHDAC5) was infused into the DG, a hippocampal subregion whose volume is reduced in major depressive disorder (MDD) [24]. Next, rats were given a CUS regimen for 4 weeks (Fig. 4A). Given that HDAC5 is expressed in the granule cell layer of the hippocampus [25], lenti-GFP was infused into DG granule cells for rats in the control groups (Fig. 4B). Lenti-shHDAC5 infusions decreased the level of HDAC5 protein as assessed by immunoblotting (Fig. 4C) and also reduced the Hdac5 mRNA level in DGs as measured by quantitative PCR (Fig. 4D). To investigate whether lentivirus infusion affected rat motor function, a locomotor activity was tested. In the LMA, there was no difference between the lenti-GFP and lenti-shHDAC5 groups (Fig. 4E). This finding indicates that lenti-GFP- and lenti-shHDAC5-infused rats had identical motor function.
 | Fig. 4HDAC5 knockdown produced antidepressant effects and occluded the actions of CPP in chronically stressed rats.(A) Schematic diagram of the experimental design. Rats were injected bilaterally with lenti-GFP or lenti-shHDAC5. (B) GFP expression in the DG (scale bar, 400 µm). (C, D) Efficiency of lentiviral-mediated knockdown of HDAC5 in the DG on the protein and mRNA levels (n=3 per group). (E) Locomotor activity, as assessed by total distance moved in the box (n=15 per group). Values are expressed as mean±S.E.M. Student's t-test. (F) Immobility times in the lenti-GFP and lenti-shHDAC5 groups at the early stage of the FST (5 min). Main effect of CPP: F1,27=5.006, p<0.05; main effect of virus: F1,27=11.678, p<0.01; interaction: F1,27=3.979, p>0.05. (G) Immobility times in the lenti-GFP and lenti-shHDAC5 groups at the late stage of the FST (10 min). Main effect of CPP: F1,27=5.496, p<0.05; main effect of virus: F1,27=9.851, p<0.01; interaction: F1,27=2.379, p>0.05. (Lenti-GFP CTL, n=7; Lenti-GFP CPP, Lenti-shHDAC5-GFP CTL, Lenti-shHDAC5-GFP CPP: n=8). (H) Bdnf mRNA level in the lenti-GFP and lenti-shHDAC5 groups (n=3 per group). Main effect of CPP: F1,12=0.582, p>0.05; main effect of virus: F1,27=13.769, p<0.01; interaction: F1,27=2.657, p>0.05. (I) BDNF protein level in the lenti-GFP and lenti-shHDAC5 groups (n=3 per group). Main effect of CPP: F1,8=5.884, p<0.05; main effect of virus: F1,8=1.278, p>0.05; interaction: F1,8=4.991, p>0.05. Two-way ANOVA was followed by LSD post hoc analysis. *p<0.05, **p<0.01 compared to lenti-GFP-CTL rats. n.s., not significant.
|
Consistent with the results shown in Fig. 3C, the immobility time during the initial 5 min period was reduced by CPP in animals infused with the lenti-GFP control virus (Fig. 4F). Rats injected with lenti-shHDAC5 showed behaviors similar to those on antidepressants. Compared to its effects in lenti-GFP-infused rats, CPP had no effects in lenti-shHDAC5-expressing rats. Since immobility in the FST was not affected (Figs. 4F and G), the antidepressant effects of CPP are likely blocked by lenti-shHDAC5 infusion. Furthermore, we found that viral-mediated hippocampal knockdown of HDAC5 induced Bdnf mRNA and protein expression, and blocked the enhancing effects of CPP on BDNF expression in stressed rats (Figs. 4H and I). Taken together, these results indicate that HDAC5 knockdown produces antidepressant effects and prevents the actions of CPP in chronically stressed animals.
Go to :
DISCUSSION
In this study, we found that CPP has antidepressant actions, and that these actions are mediated by phosphorylation of HDAC5 in the rat hippocampus. CPP rapidly induced phosphorylation of HDAC5 at Ser259 and Ser498 in the rat hippocampus. Moreover, HDAC5 phosphorylation coincided with phosphorylation of PKD and CaMKII in the hippocampus 1 h after CPP injection. The protein level of BDNF was increased by CPP and HDAC5 was less enriched at the Bdnf promoters in hippocampal neurons treated with CPP than in those cells treated with saline.
Consistent with a previous study [13], we found that CPP rapidly induced an antidepressant-like response to behavior as assessed by the FST and the NSFT. And the antidepressant-like effect of CPP can be maintained until 24 h after one administration according to the previous study [13]. These effects depend on the rapid synthesis of BDNF as a result of reduced eEF2 phosphorylation and de-suppression of translation of BDNF [13]. Our results show that HDAC5 phosphorylation could be also involved in the antidepressant actions of CPP that remain in place for at least 24 h even after CPP has been removed from receptors. Since CPP induces nuclear export of HDAC5 by increasing HDAC5 phosphorylation, CPP might regulate the transcription of genes implicated in antidepressant effects, especially those downstream of HDAC5. Considering that synaptic plasticity and dendritic morphology have been implicated in depression pathophysiology [2627], CPP has been hypothesized to increase the translation of synaptic proteins such as GluR1, synapsin1, and postsynaptic density 95 (PSD95). CPP has also been hypothesized to enhance dendritic morphology. Thus, it would be interesting to study the cellular mechanisms that underlie CPP actions to facilitate development of safer compounds with similar rapid antidepressant effects for long durations.
Many studies are currently investigating the therapeutic mechanisms of NMDA receptor antagonists for the treatment of depressive disorders and are moving beyond the monoamine system. In preclinical and clinical contexts, NMDA receptor antagonists for the treatment of depressive disorders are largely restricted to ketamine; little attention has been paid to other types of NMDA receptor antagonist. Meanwhile, it also remains unclear whether the mechanisms by which NMDA receptor antagonists trigger antidepressant-like effects are NMDA receptor engagement-independent. It has been previously reported that the antidepressant actions of ketamine metabolites are independent of NMDA receptor inhibition [28]. In line with this finding, ketamine has been shown to act on the dopamine system [29], opioid signaling [30], and the inflammatory system [31]. Taken together, these findings suggest that NMDA receptor antagonists exert their antidepressant-like effects via NMDA receptor inhibition-independent mechanisms.
Here we showed that CPP also exerts antidepressant-like effects in rats stimulated with CUS, and that these effects are mediated by phosphorylation of HDAC5. These data establish hippocampal HDAC5 as a common regulator in the molecular machinery underlying the actions of NMDA receptor antagonists.
Go to :
 XML Download
XML Download