

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ischemic stroke causes a considerable public health problem. Stroke was the second-leading global cause of death behind heart disease in 2013, accounting for 11.8 percent of total deaths worldwide [1]. Recently, it was reported that stroke ranked No. 5 among all causes of death, when considered separately from other cardiovascular diseases, and was a leading cause of serious long-term disability in the USA [2]. Advances in endovascular therapeutic approaches for patients with acute ischemic stroke, including thrombolytic therapy with tissue plasminogen activator [3] and mechanical thrombectomy [4] have led to the decline in stroke mortality [2]. Nonetheless, there is currently insufficient evidence to demonstrate the usefulness and benefit of endovascular treatment for acute ischemic stroke in patients.
A growing body of research indicates that neuroinflammation plays a key role in the pathophysiology of cerebral ischemia. Ischemic stroke can elicit a neuroinflammatory reaction in the brain within a few hours after a stroke that lasts for days or weeks as a delayed tissue reaction to injury [5]. Various inflammatory mediators, such as chemokines, cytokines, and immune cells, contribute to acute cerebral ischemic injury [6]. Postischemic inflammation is characterized by a rapid activation of resident microglial cells and by infiltration of neutrophils and macrophages in the injured parenchyma [7], as demonstrated both in animal models [89] and in stroke patients [1011]. These cells may accumulate in the capillaries, further impairing cerebral blood flow, or extravasate into the brain parenchyma. Infiltrating leukocytes, as well as resident brain cells, including neurons and glia, may release pro-inflammatory mediators, such as cytokines, chemokines and oxygen/nitrogen free radicals that contribute to the progress of tissue damage [7]. Agents that can modulate multiple pro-inflammatory mediators may provide therapeutic intervention.
Caffeic acid phenethyl ester (CAPE), an active component of the propolis purified from the hives of honeybees [12]. It has been demonstrated that CAPE has various biological and pharmacological properties, such as antioxidant [1314], anti-inflammatory [15,16] and immunomodulatory activities [1718]. However, the precise mechanism of action of CAPE to reduce the brain injury after focal cerebral ischemia is not yet established. Thus, this study was designed to investigate the protective effect of CAPE on the postischemic brain injury and neuroinflammation following photothrombotic cortical ischemia in mice.
Go to : 
METHODS
Animals
All experimental procedures were performed in accordance with the Animal Care Guidelines of the Laboratory Animal Resource Center of Pusan National University School of Medicine after receiving approval of Pusan National University Institutional Animal Care and Use Committee (PNU-2013-0424). Male C57BL/6 mice weighing 20-25 g (Koatech, Pyeongtaek, Gyeonggi-do, Korea) were housed in groups of three to five cage in a temperature-controlled environment (21-23℃, 12-h light/dark cycle) and were acclimatized in the animal facility for at least 4 days prior to use. Animals had a standard pelleted feed and water ad libitum.
Photothrombotic cortical ischemia
Permanent focal ischemia was induced by cortical photothrombotic vascular occlusion as previously described [19]. Mice were anesthetized with chloral hydrate (450 mg/kg, i.p.) and placed in a head-holding adaptor (SG-4N; Narishige, Tokyo, Japan) with a heating pad (Homeothermic blanket system; Harvard Apparatus Inc., Edenbridge, Kent, UK) placed beneath the animal to maintain the rectal temperature at 37±0.5℃. A midline scalp incision and pericranial tissue dissection revealed the bregma and lambda points. A fiber-optic bundle of a cold light source (KL 1500 LCD; Carl Zeiss, Göttingen, Germany) with a 4-mm aperture was centered using a micromanipulator at a lateral distance of 2 mm from the bregma. The aperture of the cold light source was placed as close as possible to the skull in order to avoid scattering of light. The skull was then irradiated for 15 min starting at 5 min after the i.p. injection of the photosensitive dye rose bengal (0.1 ml of 10 mg/ml; Sigma-Aldrich, St. Louis, MO, USA) in order to achieve photochemically-induced focal ischemia. Afterwards, the scalp was sutured, and the mice were allowed to awaken. The sham group was treated similarly, with the exception of irradiation and rose bengal injection. All mice tolerated the entire procedure very well, showed no visible neurological or behavioral deficits, and survived brain ischemia.
Measurement of infarct size
The infarct size, including infarct area and volume, was measured by the method previously described [19] with slight modifications. Mice were euthanized with a lethal dose of urethane (2 g/kg, i.p.) and decapitated 24 h after ischemic insult. The brain was quickly removed and chilled in ice-cold saline for 10 min. Five coronal sections (2-mm thick) were cut with a mouse brain matrix (RBM-2000C; ASI Instruments Inc., Warren, MI, USA) beginning 2 mm posterior to the anterior pole. The sections were then immersed in a saline solution containing 2% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) at 37℃ for 30 min and were fixed by immersion in a 10% neutral buffered formalin solution [20]. The dorsal surface of each section was scanned using a digital camera, and infarct area was quantified using an image analyzing system (AxioVision LE; Carl Zeiss). Infarct volume for each section was calculated by multiplying the infarct area by the section thickness. Total infarct volume in an animal was determined by summing up the infarct volumes of five sections.
Immunohistochemistry
Immunohistochemical experiments were conducted by the method previously described [21] with slight modifications. Mice were anesthetized and transcardially perfused with ice-cold 0.1 M phosphate-buffered saline (PBS; pH 7.4) containing 20 U/ml heparin followed by ice-cold 4% paraformaldehyde in PBS (pH 7.4). The brains were quickly removed, postfixed in 4% paraformaldehyde solution overnight at 4℃, and immersed in 20% sucrose until they sank. Coronal sections (7-µm thick) were cut in a cryostat at −20℃ using a microtome (HM 560; Microm International GmbH, Waldorf, Germany). The sections were adhered to poly-L-lysine-coated slides, and allowed to dry at room temperature. After quenching endogenous peroxidase in 0.6% H2O2 and blocking non-specific protein-binding with CAS-Block (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min, the preparations were incubated overnight at 4℃ with the primary antibodies, including mouse monoclonal anti-tumor necrosis factor-α (anti-TNF-α; 1:200; sc-52746, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), rabbit anti-hypoxia-inducible factor-1α (anti-HIF-1α; 1:100; A300-286A, Bethyl Laboratories, Inc., Montgomery, TX, USA), rat monoclonal anti-monocyte chemoattractant protein-1 (anti-MCP-1; 1:150; Abcam, Cambridge, MA, USA), rabbit anti-heme oxygenase-1 (anti-HO-1; 1:100; SPA-895, Stressgen Biotechnologies Co., Victoria, BC, Canada), and rabbit anti-mouse anti-indoleamine 2,3-dioxygenase (anti-IDO; 1:200; ALX-210-432-C100, Enzo Life Sciences, Inc., Farmingdale, NY, USA). The sections were washed with PBS, and then incubated for 3 h with the appropriate secondary antibodies, such as biotinylated anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA), biotinylated anti-rat IgG (Stressgen), and biotinylated anti-mouse IgG (Stressgen) at a dilution of 1:100. After washing three times with PBS, the slides were incubated for 1 h with avidin and biotinylated horseradish peroxidase complex (VECTASTAIN Elite ABC kit, Vector Laboratories). They were then washed in PBS, incubated with diaminobenzidine substrate kit (Vector Laboratories) and rinsed in water. The cortical ischemic core region was examined for positive staining of TNF-α, HIF-1α, MCP-1, HO-1, and IDO by light microscopy.
Western blot analysis
Western blotting experiments were conducted by the method previously described [19] with slight modifications. The ipsilateral cerebral cortex including both ischemic core and penumbra regions was used as a sample for Western blot analysis. Samples were homogenized in an ice-cold lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA; 1% Triton X-100; 10% glycerol; 100 µg/ml phenylmethylsulfonyl fluoride; 1 mM sodium orthovanadate; and 5 mM sodium fluoride). Following centrifugation at 14,000 g for 7 min, 50 µg of total protein from each sample was loaded into a 10% SDS-polyacrylamide electrophoresis gel and transferred onto a nitrocellulose membrane (Amersham Protran Premium 0.45 NC; GE Healthcare Bio-Sciences Corp., Pittsburgh, PA, USA). After blocking with non-fat milk, the membrane was incubated with the following primary antibodies: mouse anti-TNF-α (1:200, Santa Cruz), mouse monoclonal anti-HIF-1α (Clone H1α67) (1:1,000; Cay-10347; Cayman Chemical, Ann Arbor, MI, USA), rabbit anti-HO-1 (1:1000; Stressgen), rabbit anti-IDO (1:2000, Enzo), and mouse monoclonal anti-β-actin (1:1,000; Santa Cruz). The immunoreactive bands were detected using the chemiluminescent reagent of the SuperSignal West Dura Extended Duration Substrate Kit (Thermo Fisher Scientific Pierce Biotechnology, Rockford, IL, USA). The band intensities were determined using GS-710 Calibrated Imaging Densitometer (Bio-Rad Laboratories, Inc., Hercules, CA, USA), and were expressed as the relative ratio to β-actin.
Enzyme-linked immunosorbent assay (ELISA)
The ipsilateral cerebral cortex including both ischemic core and penumbra regions was used as a sample for the ELISA. Samples were homogenized in the lysis buffer: 200 mM NaCl, 5 mM EDTA, 10 mM Tris, 10% glycerol, and 1 mM phenylmethylsufonyl fluoride. Samples were then centrifuged twice (14,000 g at 4℃ for 7 min) to avoid contamination with cell debris, and the cleared supernatants were used for the ELISA. The levels of interleukin-1α (IL-1α) and interleukin-10 (IL-10) were determined using ELISA kits for mouse IL-1α and mouse IL-10 (KOMA Biotech, Inc., Seoul, Korea) as per the manufacturer's instructions, respectively.
Drug administration
Mice were randomly assigned to groups in random order. CAPE (Sigma-Aldrich) was dissolved in 0.3% dimethyl sulfoxide, and was administered i.p. twice in doses from 0.5 mg/kg to 5 mg/kg 1 h and 6 h after ischemic insult in a volume of 1 ml/100 g.
Statistical analysis
All data are expressed as mean±SEM. The statistical differences in mean values between drug-treated and vehicle groups were analyzed by one-way analysis of variance followed by Tukey's multiple comparison test as a post hoc test using a statistical software (Prism, version 5.01; GraphPad Software Inc., San Diego, CA). A value of p<0.05 was considered statistically significant.
Go to :
RESULTS
Infarct size
Ischemic insult markedly increased the infarct size, including infarct area and volume, both of which were reduced by CAPE treatment (Fig. 1). CAPE significantly reduced the infarct area in the second section at a dose of 2 mg/kg (p<0.01) and in the first, second, and third sections at a dose of 5 mg/kg (p<0.05, p<0.01, and p<0.05, respectively; Fig. 1B), compared to the corresponding value in the vehicle group. This resulted in a significant reduction in the total infarct volume (p<0.05 at 2 mg/kg and p<0.01 at 5 mg/kg, respectively; Fig. 1C).
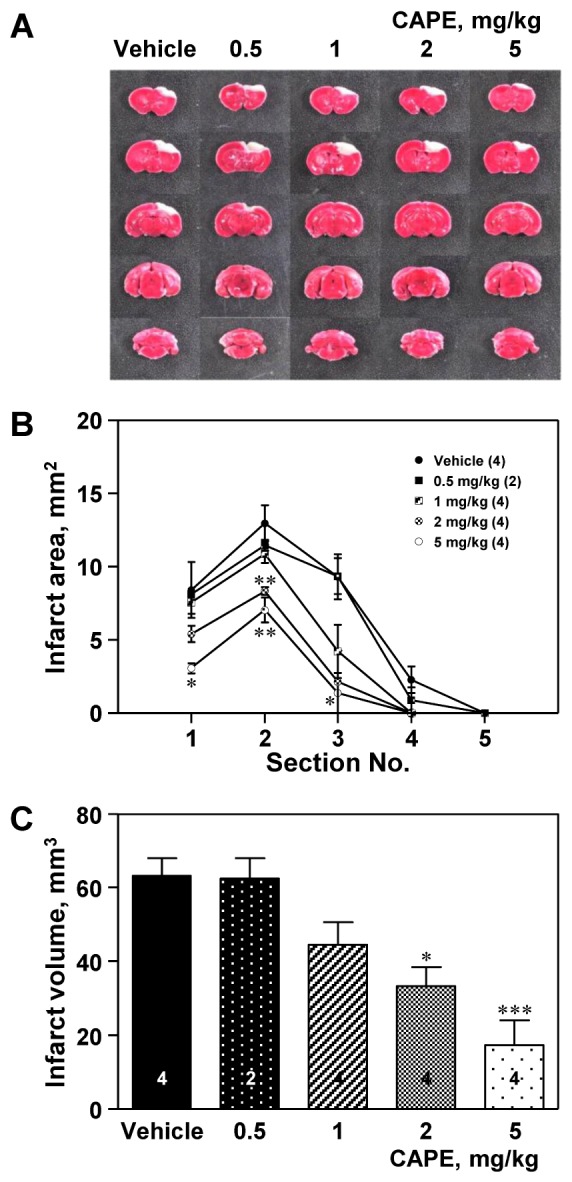 | Fig. 1Effect of caffeic acid phenethyl ester (CAPE) on infarct size.(A) Representative photographs showing the ischemic lesion. Male C57BL/6 mice were treated with CAPE (0.5–5 mg/kg, i.p.) twice 1 and 6 h after ischemic insult, and were sacrificed 24 h after photothrombotic cortical ischemia. The pale non-stained area indicates the lesion of ischemic injury, while the deep red-colored area indicates the viable tissue. CAPE elicited the reduction in the infarct area (B) and infarct volume (C). The numbers in parentheses and columns indicate the numbers of animals. Data are presented as mean±SEM. *p<0.05; **p<0.01; ***p<0.001 vs. vehicle group.
|
Expression of TNF-α
The number of TNF-α positive cells increased in the cortical ischemic core region ipsilateral to the photothrombosis compared to that in the sham-operated group (Fig. 2). CAPE treatment (5 mg/kg) significantly reduced the number of TNF-α positive cells compared to the vehicle group (79.4±11.8 positive cells/section vs. 121.4±10.5 positive cells/section, p<0.05; Fig. 2A). Western blot analysis revealed that ischemic insult markedly increased TNF-α protein expression, which was significantly reduced by treatment with CAPE 5 mg/kg (p<0.001; Fig. 2B).
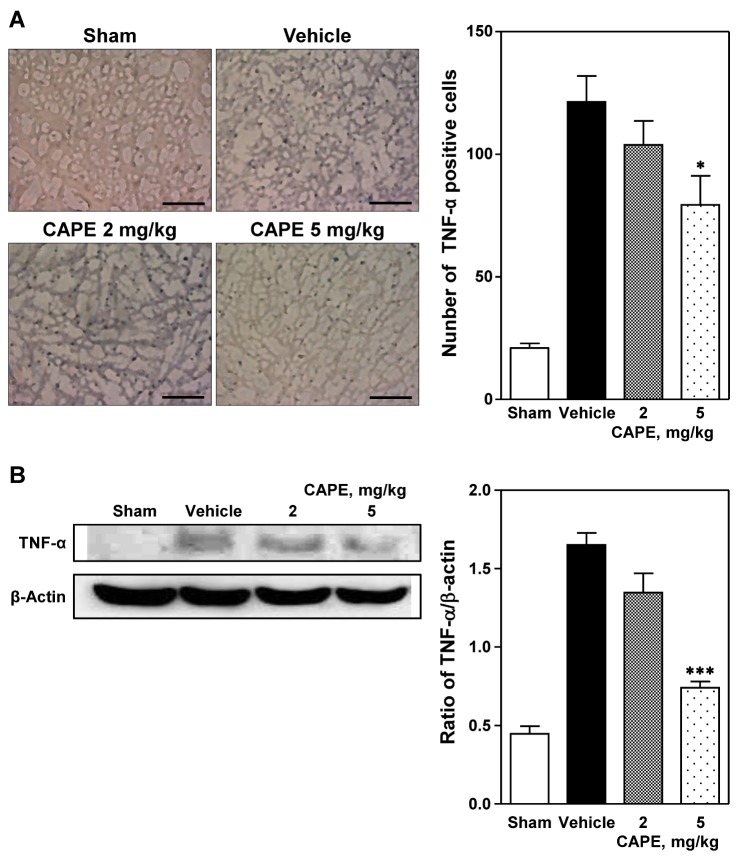 | Fig. 2Immunohistochemical and Western blot analyses of expression of tumor necrosis factor-α (TNF-α).(A) Representative photomicrographs showing TNF-α positive cells (left panel) and number of TNF-α positive cells (right panel) in the cortical ischemic core region 24 h after photothrombosis. Treatment with CAPE 5 mg/kg significantly reduced the immunohistochemical staining of TNF-α positive cells compared to the vehicle group. Five animals were used in each group. Scale bar=100 µm. (B) Western blot analysis revealed that TNF-α expression was reduced by CAPE in a dose-dependent manner. Data are presented as mean±SEM. *p<0.05; ***p<0.001 vs. vehicle group.
|
Expression of HIF-1α and MCP-1
Immunohistochemical analysis revealed a marked increase in the expression of HIF-1α and MCP-1 in the cortical ischemic core region ipsilateral to the photothrombosis compared to that in the sham-operated group (Figs. 3A and B, respectively). Treatment with CAPE 5 mg/kg significantly reduced the number of HIF-1α positive cells in a dose-dependent manner, compared to the vehicle group (124.7±9.4 positive cells/section vs. 188.0±6.3 positive cells/section, p<0.001; Fig. 3A) [F(2,15)=17.0, p<0.001]. In addition, CAPE significantly reduced the number of MCP-1 positive cells (142.3±9.8 and 93.5±14.6 positive cells/section at 2 and 5 mg/kg; p<0.05 and p<0.001, respectively) in a dose-dependent manner, compared to the vehicle group (188.7±6.4 positive cells/section; Fig. 3B) [F(2,15)=19.4, p<0.001].
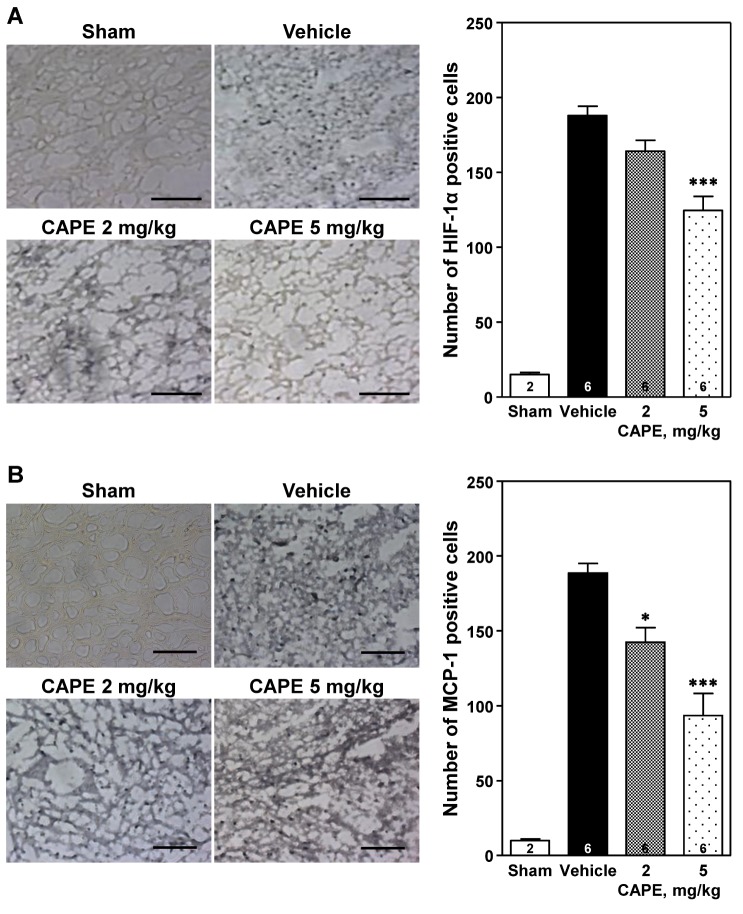 | Fig. 3Immunohistochemical analysis of expression of hypoxia-inducible factor-1α (HIF-1α) and monocyte chemoattractant protein-1 (MCP-1).Representative photomicrographs showing HIF-1α and MCP-1 positive cells (left panels of A and B, respectively) and number of HIF-1α and MCP-1 positive cells (right panel panels of A and B, respectively) in the cortical ischemic core region 24 h after photothrombosis. Treatment with CAPE dose-dependently reduced the immunohistochemical staining of HIF-1α and MCP-1 positive cells compared to the vehicle group. Scale bar=100 µm. The numbers in columns indicate the numbers of animals. Data are presented as mean±SEM. *p<0.05; ***p<0.001 vs. vehicle group.
|
Expression of HO-1
The number of HO-1 positive cells increased in the cortical ischemic core region ipsilateral to the photothrombosis compared to that in the sham-operated group (Fig. 4). CAPE dose-dependently increased the number of HO-1 positive cells [F(2,15)=16.5, p<0.001], with a statistical significance at doses of 2 and 5 mg/kg (181.5±4.8 and 207.3±6.1 positive cells/section at 2 and 5 mg/kg vs. 149.2±9.7 positive cells/section; p<0.05 and p<0.001, respectively).
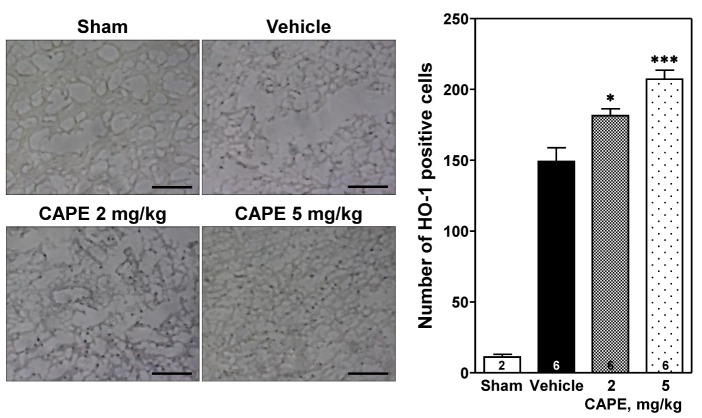 | Fig. 4Immunohistochemical analysis of expression of heme oxygenase-1 (HO-1).Representative photomicrographs showing HO-1 positive cells (left panel) and number of HO-1 positive cells (right panel) in the cortical ischemic core region 24 h after photothrombosis. CAPE dose-dependently increased the number of HO-1 positive cells compared to the vehicle group. Scale bar=100 µm. The numbers in columns indicate the numbers of animals. Data are presented as mean±SEM. *p<0.05; ***p<0.001 vs. vehicle group.
|
Expression of IDO
The number of IDO positive cells increased in the cortical ischemic core region ipsilateral to the photothrombosis compared to that in the sham-operated group (Fig. 5). Treatment with 5 mg/kg of CAPE significantly reduced the number of IDO positive cells compared to the vehicle group (90.0±12.3 positive cells/section vs. 166.8±19.1 positive cells/section, p<0.01; Fig. 5A). Western blot analysis revealed that ischemic insult markedly increased IDO protein expression, which was significantly reduced by treatment with 5 mg/kg of CAPE (p<0.001; Fig. 5B).
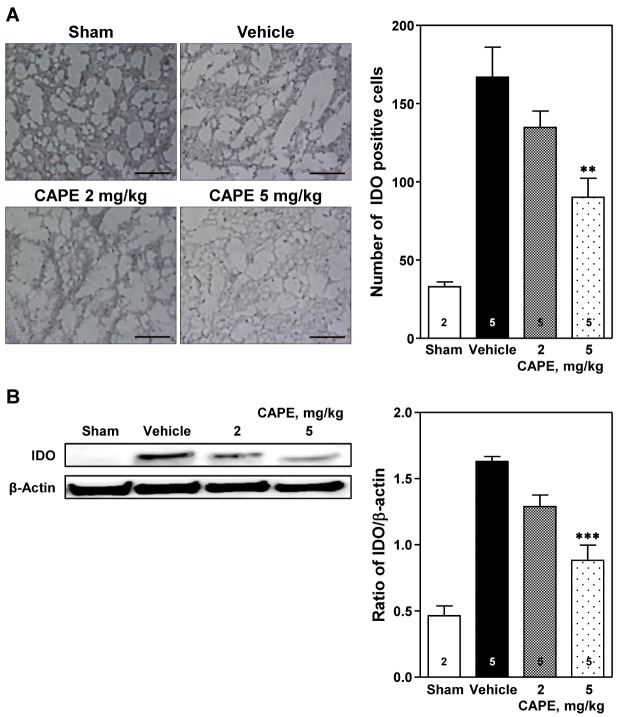 | Fig. 5Immunohistochemical and Western blot analyses of expression of indoleamine 2,3-dioxygenase (IDO).(A) Representative photomicrographs showing IDO positive cells (left panel) and number of IDO positive cells (right panel) in the cortical ischemic core region 24 h after photothrombosis. Treatment with CAPE 5 mg/kg significantly reduced the immunohistochemical staining of IDO positive cells compared to the vehicle group. Scale bar=100 µm. (B) Western blot analysis revealed that IDO expression was significantly reduced by CAPE 5 mg/kg. The numbers in columns indicate the numbers of animals. Data are presented as mean±SEM. **p<0.01; ***p<0.001 vs. vehicle group.
|
Levels of IL-1α and IL-10
Ischemic insult markedly increased the production of IL-1α in the cerebral cortex ipsilateral to the photothrombosis as compared to that in the sham-operated group. CAPE dose-dependently decreased the level of IL-1α [F(3,12)=22.2, p<0.001; Fig. 6, left panel], with a statistical significance at doses of 2 and 5 mg/kg (p<0.001 and p<0.001, respectively). However, ischemic insult did not affect IL-10 level in the cerebral cortex ipsilateral to the photothrombosis. CAPE dose-dependently increased IL-10 levels [F(3,12)=326.7, p<0.001; Fig. 6, right panel], with a statistical significance at 2 and 5 mg/kg (p<0.001 and p<0.001, respectively).
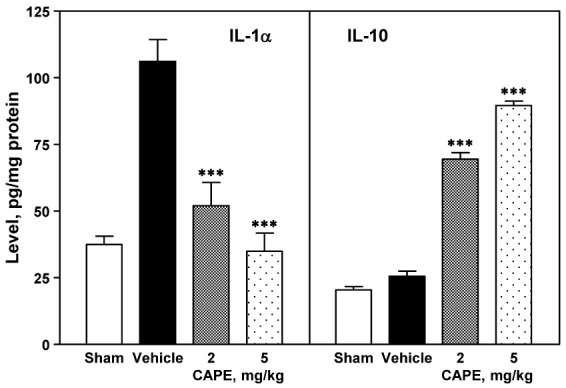 | Fig. 6Effect of CPAE on production of IL-1α and IL-10.The cerebral cortex ipsilateral to the photothrombosis was obtained 24 h after photothrombotic cortical ischemia, and was homogenized in lysis buffer. The supernatant of centrifuged sample was used for measuring the levels of IL-1α and IL-10 using ELISA (left and right panels, respectively). CAPE reduced the production of IL-1α (left panel) and enhanced production of IL-10 (right panel) in the ischemic region in a dose-dependent manner. Four animals were used in each group. ***p<0.001 vs. vehicle group.
|
Go to :
DISCUSSION
In the present study, we examined the effects of CAPE on photothrombotic cortical ischemia in mice. Postischemic treatment with CAPE reduced infarct size via induction of anti-inflammatory mediators as well as reduction in pro-inflammatory mediators within the cerebral cortex ipsilateral to the photothrombosis.
CAPE, a natural flavonoid-like compound, is one of the major components of honeybee propolis [12]. Propolis has been used for centuries in folk medicine [22], and to date no harmful effects of CAPE have been detected in normal cells [23]. Furthermore, CAPE appears to exhibit antioxidant [1314], anti-inflammatory [1516], anti-viral [24], anti-carcinogenic [25], and immunomodulatory properties [1718]. It has also been reported that CAPE treatment protects tissues against ischemia-reperfusion injury [2326], and oxidative damages [27]. In the present study, CAPE-treated brain sections revealed that the pale non-stained area was significantly reduced, compared to the vehicle group. This result of reduction in infarct size involves demonstrating a protective effect of CAPE against ischemia.
Following photothrombotic focal cerebral ischemia, the volume of ischemic damage reaches maximal size 24 h after ischemia [28]. The largest increment of infarct volume occurs during the subacute phase in which the infarct core expands into the penumbra until, after 4–6 h, it becomes congruent with it. After 3 h, more than 50% and between 6 and 8 h, almost all of the penumbra disappears and becomes part of the irreversibly damaged infarct core [29]. Although the penumbral region is generally considered salvageable and is the main pharmacological target for acute ischemic stroke treatment [30], in the current study, we examined the effect of CAPE on expression of immunohistochemically positive pro- and anti-inflammatory mediators in the cortical ischemic core region where they dominantly express and ultimately cause irreversible neuronal injury immediately after ischemia [3132].
In ischemic stroke condition, upregulaton of inflammatory mediators happens from brain-resident cells and infiltrating immune cells [33]. Neuroinflammation within the brain is associated with a number of pro- and anti-inflammatory cytokines/chemokines/transcription factors/enzymes [3334]. TNF-α, a principal mediator of neuroinflammation, is induced very early in the injury process [3536], and elicits a cascade of cellular events culminating in neuronal death [37]. In addition, the present study confirmed that the photothrombotic cortical ischemia markedly elevated the level of TNF-α, which were significantly reduced by CAPE treatment, indicating that CAPE functions as an attenuator of TNF-α expression in the cerebral cortex ipsilateral to the photothrombosis.
HIF-1α is a key component of the cellular response to hypoxia and ischemia under pathophysiologic conditions, such as stroke [38]. It can modulate ischemic injury via induction of target genes that may protect or destroy ischemic neurons [39]. The neuroprotective effect of HIF-1α and its benefit target genes [4041] has been widely examined in preconditioning or after the ischemic phase [42]. In contrast, in severe hypoxic cases, HIF-1α gets accumulated and leads to cell death or apoptosis by mediating different target genes [384344]. HO-1, an inducible form of HO, has been demonstrated to be regulated by HIF-1α [4546]. A growing body of evidence has suggested that HO plays an important role in protection against cerebral ischemia, confirming that induction of the HO-1 protein expression following cerebral ischemia can lead to a beneficial outcome [4748]. The present study showed that CAPE significantly inhibited HIF-α expression, therefore, CAPE treatment might protect against brain damage following focal cerebral ischemia. However, the expression level of HO-1 was increased dose-dependently by treatment with CAPE. This opposing effect of CAPE on HIF-1α and HO-1 expression is supported by the report of Demougeot et al. [49] that HIF-independent mechanisms prevail over HIF-dependent mechanisms in HO-1 induction after brain ischemia.
In the present study, CAPE significantly and dose-dependently reduced the number of MCP-1 positive cells in the cortical ischemic core region. It has been reported that elevated MCP-1 level is responsible for the increased influx of inflammatory cells, which in turn leads to sustained brain damage after cerebral infarction [50]. MCP-1 deficiency attenuates infarct volume in mice [51], however, overexpression of MCP-1 resulted in larger infarcts and increased chemoattraction of monocytes and macrophages into the ischemic area compared to that in wild-type animals [52]. TNF-α induces MCP-1 expression, leading to macrophage recruitment [53] and upregulation of MCP-1 secretion in the brain [54]. Based on these reports, it could be speculated that upregulation of MCP-1 in the cortical ischemic core region was induced by elevated TNF-α level following cerebral ischemia, and this change was also attenuated by CAPE treatment. However, the interrelation between TNF-α and MCP-1 is obscure in the present study, and the exact mechanism underlying the effect of CAPE on cerebral ischemia-induced MCP-1 upregulation remains to be clarified.
IDO is the first and rate-limiting enzyme in the generation of quinolinic acid from L-tryptophan via the kynurenine pathway outside of the liver [55], and is highly induced under cerebral ischemia [56]. IDO has been implicated in the pathogenesis of inflammatory neurologic diseases such as ischemic brain disease [57]. The increased production of proinflammatory cytokines (such as IL-1β, TNF-α and even IL-18) resulting from stroke leads to the amplification of the inflammatory processes and widespread activation of the IDO enzyme [58]. In the present study, induction of IDO in the ipsilateral cortex was associated with focal cerebral ischemia, which was significantly suppressed by CAPE treatment. To our knowledge, this is the first study to investigate the reduction in IDO expression in the cerebral cortex ipsilateral to the photothrombosis following CAPE treatment. Future studies are needed to elucidate the mechanism underlying the attenuation of IDO expression within the ischemic brain by CAPE.
IL-1 is a pro-inflammatory cytokine that has been identified as an important mediator of neurodegeneration induced by experimental cerebral ischemia or excitatory or traumatic brain injury in rodents [5960]. Although both IL-1α and IL-1β are induced in response to cerebral ischemia [59], IL-1α is expressed as early as 4 h after reperfusion following ischemia induced by occlusion of the middle cerebral artery. In addition, it is the major form of IL-1 contributing to inflammation early after cerebral ischemia [61]. In the present study, CAPE dose-dependently decreased IL-1α levels in the cerebral cortex ipsilateral to the photothrombosis. It can be suggested that postischemic treatment with CAPE 1 and 6 h after ischemic insult attributes to the decrease in IL-1α production. IL-10 is a major anti-inflammatory cytokine produced by various inflammatory cell types, particularly macrophages. It is believed to protect against ischemic brain damage via its anti-inflammatory and anti-apoptotic properties [6263]. In the present study, CAPE dose-dependently increased IL-10 levels in the cerebral cortex ipsilateral to the photothrombosis. This finding demonstrates that increased IL-10 level induced by CAPE treatment contributes to the reduction of ischemic brain damage. It can be predicted that CAPE provides a therapeutic potential for the prevention of cerebral ischemic damage by reducing inflammatory processes via increasing IL-10 production as well as reducing IL-1α production in the brain subjected to focal cerebral ischemia.
In the present study, we mainly observed a direct neuroprotective effect of CAPE on postischemic infarct size and expression of pro- and anti-inflammatory mediators in the cerebral cortex ipsilateral to the photothrombosis. However, we did not assess the effectiveness of CAPE in morphofunctional recovery after photothrombotic cortical ischemic injury. In addition, we did not assess the discriminatory effect of CAPE on the infarct core and ischemic penumbra at the cellular and molecular level, respectively. Further studies are required to evaluate the neuroprotective effect of CAPE using methods for knockdown or knockout of pro-inflammatory genes and to identify the cell types exhibiting immunoreactivity for pro- and anti-inflammatory mediators. Moreover, the association between anti-inflammatory properties of CAPE and morphofunctional improvement needs to be clarified in future studies.
In summary, posttreatment with CAPE significantly reduced the infarct size, and dose-dependently reduced the expression of TNF-α, HIF-1α, MCP-1, and IDO as well as the production of IL-1α in the cerebral cortex ipsilateral to the photothrombosis. In addition, CAPE elicited a significant increase in HO-1 expression and IL-10 production in the cerebral cortex ipsilateral to the photothrombosis. Taken together, these findings suggest that CAPE exerts a remarkable neuroprotective effect on ischemic brain injury via its anti-inflammatory properties, thereby providing a benefit to the therapy of cerebral infarction.
Go to :
 XML Download
XML Download