

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammation, an innate immune response mediated mainly by macrophages, is a series of biological processes to protect the body from invading pathogens including bacteria, viruses, protozoans, and fungi. Inflammation is characterized by pain, heat, swelling, redness, and loss of function [123]. An inflammatory response is initiated by recognition of extracellular pathogen-associated molecular patterns (PAMPs), which are pathogenic components derived from invading pathogens through their molecular receptors called pattern recognition receptors (PRRs) expressed on the cell surfaces of macrophages [134]. One of the most intensively studied PRRs is Toll-like receptor 4 (TLR4), which is the molecular receptor for extracellular lipopolysaccharide (LPS), one of the most pathogenic components derived from the cell walls of Gram-negative bacteria [5]. Once extracellular LPS is recognized by TLR4 on macrophages, a macrophage-mediated inflammatory response is immediately induced. Inflammatory signaling pathways including nuclear factor-kappa B (NF-κB), activator protein-1 (AP-1), and interferon regulatory factors (IRFs) pathways are activated by inducing the signal transduction cascades of intracellular inflammatory signaling molecules. The activated inflammatory signaling pathways not only up-regulate expression of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), and IL-6, and inflammatory genes including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), but also increase the secretion of inflammatory mediators including nitric oxide (NO) and prostaglandin E2 (PGE2) [4678].
Recent studies have demonstrated that inflammatory responses are also strongly induced by intracellular inflammasomes, protein complexes that induce inflammatory responses in macrophages by activating gasdermin-D (GSDMD)-mediated pyroptosis and the secretion of pro-inflammatory cytokines including IL-1β and IL-18 in a caspase-1-dependent manner. Inflammasomes are classified in ‘canonical inflammsomes’, such as nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and absent in melanoma 2 (AIM2) inflammasomes as well as ‘non-canonical inflammasomes’, such as caspase-4, -5, and -11 [910111213]. These inflammsomes are activated by different types of ligands, leading to the induction of inflammatory responses [910111213].
Inflammation is a host defense mechanism to remove invading pathogens from the body. However, chronic inflammation is prolonged with repeated inflammatory responses. It can last for weeks to even years and is characterized by cycles of tissue injury and healing, consequently resulting in severe tissue damage. More critically, chronic inflammation has been posited as a major risk factor for the development of inflammatory/autoimmune diseases that are various arrays of disorders or conditions characterized by chronic inflammation against self-tissues [14151617]. Rheumatic diseases are chronic inflammatory/autoimmune and degenerative diseases that mainly but not exclusively affect connective tissues, such as bones and cartilages in joints, tendons, ligaments, and muscles resulting in substantial morbidity and mortality. Since rheumatic diseases does not affect only connective tissues but some of them cause severe damages at other non-connective tissues and internal organs, there are more than 100 rheumatic diseases that are the biggest population in the inflammatory/autoimmune diseases with a large number of patients all over the world, and extensive studies have been focusing on these diseases. Many rheumatic diseases are caused by chronic inflammation and autoimmunity which are called ‘inflammatory/autoimmune rheumatic diseases’, and these diseases include rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), ankylosing spondylitis (AS), and Sjögren's syndrome (SS). Although the exact causes of rheumatic diseases are not fully understood, it is clear that chronic inflammatory responses are one of the major causative factors for development of rheumatic diseases. Therefore, it is reasonable that inflammasome-induced inflammatory responses in macrophages correlate with the development of inflammatory/autoimmune rheumatic diseases.
This review provides a general introduction to inflammasomes in macrophage-mediated inflammatory responses and discusses recent research on the role of inflammasomes in inflammatory/autoimmune rheumatic diseases. The aim of the review is to increase understanding of the inflammasome role in the pathogenesis of inflammatory/autoimmune rheumatic diseases. Insight on inflammasomes might contribute to the development of novel and promising anti-inflammatory drugs for the prevention and treatment of inflammatory/autoimmune rheumatic diseases.
Go to : 
STRUCTURES AND ACTIVATION OF INFLAMMASOMES
Inflammatory responses are initially induced by PRRs in response to a variety of extracellular and intracellular PAMPs and stimuli. Several families of intracellular PRRs, such as NLRs, leucine-rich repeats (LRRs), AIM2, retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and caspase-11 have been identified [91113181920212223242526]. Interestingly, some intracellular PRRs, such as NLRs and AIM2, induce a unique inflammatory response. These intracellular PRRs assemble protein complexes called ‘canonical inflammasomes’ consisting of a PRR, bipartite adaptor molecule, ASC, and pro-caspase-1. Different types of NLRs, such as NLRP1, NLRP3, and NLRC4, as well as the non-NLR family pathogen receptor AIM2 have been identified, and these pathogen receptors are activated by different stimuli [911]. These canonical inflammasomes activate an inflammatory caspase, caspase-1, resulting in the maturation and secretion of pro-inflammatory cytokines IL-1β and IL-18 as well as pyroptosis, an inflammatory form of programmed cell death that occurs with pathogen infection [911252728]. Recent studies have demonstrated that, caspase-11 directly recognizes intracellular LPS derived from Gram-negative bacteria and can activate inflammatory responses by inducing pyroptosis and secretion of IL-1β and IL-18 in macrophages [24]. This caspase-11-mediated inflammatory response is similar to NLRs- and AIM2-inflammasome-mediated inflammatory responses, but different from these canonical inflammasomes in composition and molecular mechanism during macrophage-mediated inflammatory responses. Therefore, this caspase-11 scaffold is considered a ‘non-canonical inflammasome’ [2425262930313233].
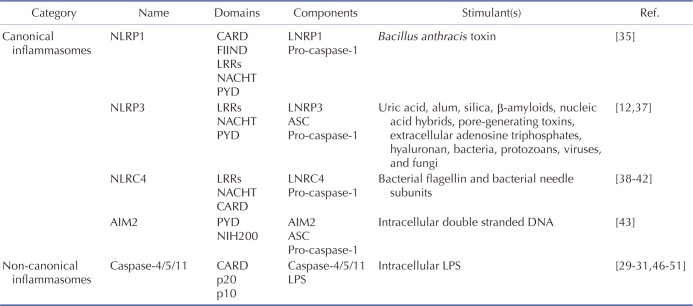
NLRP1 inflammasome is a protein complex consisting of NLRP1, which was identified as the first NLR family member to form inflammasomes [34]; ASC, which is an adaptor molecule; and pro-caspase-1 [911]. NLRP1 is the only member of its family present in humans and has an amino-terminal pyrin domain (PYD), a nucleotide-binding and oligomerization domain (NACHT), leucine-rich repeats (LRRs), a functional-to-find domain (FIIND), and a carboxyl-terminal caspase recruit domain (CARD) (Fig. 1A). Unlike human NLRP1, several isoforms of mouse NLRP1, including NLRP1A, NLRP1B, and NLRP1C, have been identified. Interestingly, a PYD motif that exists in human NLRP1 is absent in the mouse NLRP1 family (Fig. 1A) [11]. Under the stimulation of Bacillus anthracis toxin, NLRP1 forms a NLRP1 inflammasome by direct interaction with procaspase-1 through their CARD motifs in macrophages (Fig. 1A) [35] and is subsequently activated to induce pyroptosis and caspase-1-mediated secretion of IL-1β and IL-18. The critical role of NLRP1 inflammasome in macrophage-mediated inflammatory responses is supported by studies reporting that pyroptosis and caspase-1-mediated secretion of IL-1β and IL-18 are abolished in macrophages isolated from Nlrp1 knock-out (KO) mice [3536].
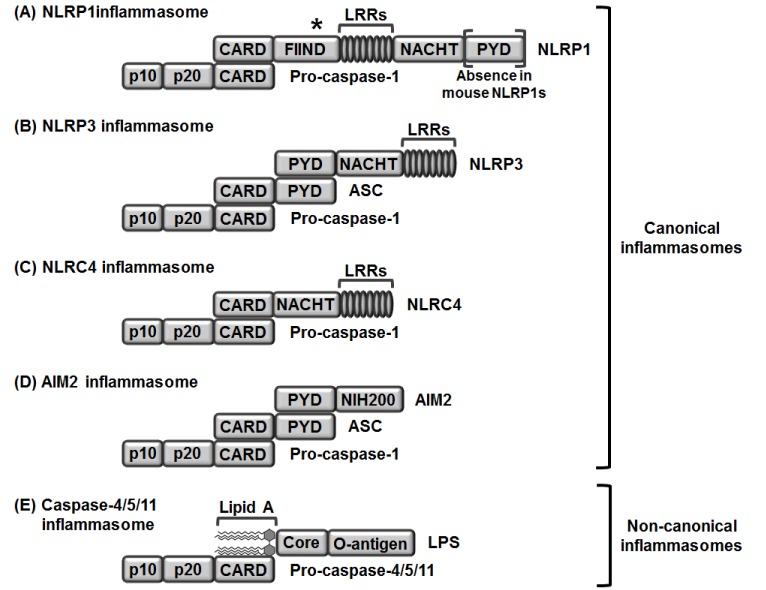 | Fig. 1Structures and compositions of canonical (A-D) and non-canonical (E) inflammasomes.(A) NLRP1 directly interacts with pro-caspase-1 through their CARD motifs. (B) NLPR3 interacts with pro-caspase-1 through a bipartite adaptor molecule, ASC. NLRP3 interacts with ASC through their PYD motifs, and ASC interacts with pro-caspase-1 through their CARD motifs. (C) NLRC4 directly interacts with pro-caspase-1 through their CARD motifs. (D) AIM2 interacts with pro-caspase-1 through a bipartite adaptor molecule, ASC. AIM2 interacts with ASC through their PYD motifs, and ASC interacts with pro-caspase-1 through their CARD motifs. (E) Pro-caspase-4/5 in a human and pro-caspase-11 in a mouse directly interact with the lipid A moiety of LPS through their CARD motifs. LRR, Leucine-rich repeat; NRL, nucleotide-binding oligomerization domain-like receptor; caspase, cysteine-aspartic protease; CARD, caspase recruit domain; NACHT, nucleotide binding and oligomerization domain; FIIND, function to find domain; AIM2, absent in melanoma 2; PYD, pyrin domain; HIN, hematopoietic interferon-inducible nuclear proteins; LPS, lipopolysaccharide. *Autocatalytic cleavage.
|
NLRP3 consists of three major parts – an amino-terminal PYD motif, a NACHT motif, and carboxy-terminal LRRs (Fig. 1B). NLRP3 responds to a number of stimuli including uric acid, alum, silica, β-amyloids, nucleic acid hybrids, pore-generating toxins, extracellular adenosine triphosphates, hyaluronan, and various pathogens including bacteria, protozoans, viruses, and fungi [1237]. Under stimulation with these ligands, NLRP3 forms an inflammasome by binding with bipartite adaptor molecule ASC through its PYD motif and recruiting and binding with procaspase-1 through its CARD motif (Fig. 1B).
NLRC4 was initially discovered as an apoptotic-protease activating factor 1 (APAF1) due to structural similarity and is very similar to NLRP3 in structure but has a CARD motif at an amino-terminus instead of PYD (Fig. 1C). NLRC4 is also stimulated by bacterial ligands, such as bacterial flagellin and bacterial needle subunits [3839404142]. In response to this stimulation, NLRC4 forms an inflammasome by directly binding with pro-caspase-1 through CARD motifs (Fig. 1C). Unlike NLRP3, NLRC4 does not need an adaptor molecule ASC to interact with pro-caspase-1.
AIM2 was identified as a direct sensor to detect intracellular double-stranded nucleic acids derived from invading pathogens and is different from NLRs due to the experimental observation that intracellular double-stranded nucleic acids from invading pathogens activate caspase-1, but not other inflammation effector molecules such as NLRs and TLRs [43]. Therefore, AIM2 was regarded as another type of intracellular receptor to trigger macrophage-mediated innate immune responses. As previously discussed, NLRP1, NLRP3, and NLRC4 are NLR family members, while AIM2 does not belong to this family, but is a p200 protein family member. AIM2 consists of two major parts – an amino-terminal PYD and a carboxy-terminal hematopoietic IFN-inducible nuclear protein (HIN) domain (Fig. 1D). In response to intracellular nucleic acids derived from pathogens, including Francisella tularensis, cytomegalovirus, and vaccinia virus, AIM2 assembles an AIM2 inflammasome by binding with bipartite adaptor molecule ASC through PYD motifs and recruiting procaspase-1 to bind with CARD motifs (Fig. 1D) [4445].
A non-canonical inflammasome was unexpectedly identified during the study of toxin-mediated inflammasome activation in macrophage-mediated inflammatory responses. Cholera toxin B (CTB) highly activated NLRP3 inflammasome and induced both pyroptosis and the secretion of IL-1β and IL-18. This inflammatory response was abolished in macrophages isolated from 129S6 mouse strain, which expresses a truncated and non-functional caspase-11 due to a polymorphism in the caspase-11 gene locus [24]. This observation strongly indicated that caspase-11, which is not a component of canonical inflammasomes, is a novel intracellular sensor that induces macrophage-mediated inflammatory responses and is different from the previously identified canonical inflammasomes and therefore named a ‘non-canonical inflammasome.’ Inactive pro-caspase-11 recognizes and directly binds with intracellular LPS, the most pathogenic component of Gram-negative bacteria, including E. coli, S. typhimurium, C. rodentium, L. pneumophilia, and Burkholderia spp., S. flexneri [293031464748495051], and the binding between these two molecules is accomplished by direct interaction of the CARD motif of pro-caspase-11 with the lipid A moiety of LPS (Fig. 1E) [32]. Caspase-4 and caspase-5 are regarded as human homologues of mouse caspase-11. Similar to mouse caspase-11, their pro-forms pro-caspase-4 and pro-caspase-5 directly bind with the lipid A moiety of intracellular LPS through CARD motifs (Fig. 1E) [325253]. Once these caspases-4/5/11 bind with intracellular LPS released from Gram-negative bacteria, caspase-4/5/11 non-canonical inflammasomes are activated by oligomerization of the caspase-4/5/11 monomers through their CARD motifs [25] and subsequently induce pyroptosis and the secretion of IL-1β and IL-18 by generating hollow-shaped pores composed of amino-terminal fragments of gasdermin D within macrophage membranes [2526335455565758]. Inflammasomes discussed here are summarized in Table 1.
Go to :
ROLES OF INFLAMMASOMES IN INFLAMMATORY/AUTOIMMUNE DISEASES
Activation of inflammasomes is an essential step to induce inflammatory responses in macrophages, but chronic inflammatory response is a major risk factor for the development of inflammatory/autoimmune diseases. This indicates that uncontrolled and repeated activation of inflammasomes could play a critical role in the pathogenesis of inflammatory/autoimmune diseases. In this section, we discuss the role of inflammasomes in the inflammatory/autoimmune rheumatic diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), ankylosing spondylitis (AP), and Sjögren's syndrome.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a serious lifelong autoimmune disease that primarily affects joints, is characterized by chronic synovial inflammation causing cartilage degradation and joint destruction, and has poor outcomes and limited treatment options [59606162]. Since RA is the inflammatory/autoimmune disease with the highest prevalence worldwide, ranging from 0.5 to 1% of the population [63], the role of inflammasomes in RA has been actively investigated.
The relationship between inflammasomes and RA pathogenesis has been explored by genetic studies. Gene expression of NLRP3 inflammasome components was analyzed in RA patients receiving infliximab, an anti-TNF monoclonal antibody therapeutic. Gene expression of NLRP3 inflammasome components, including ASC, full-length NLRP3, short length NLRP3, and caspase-1, was significantly higher in RA patients compared to controls, and single-nucleotide polymorphisms (SNPs) at NLRP3 locus were associated with RA susceptibility and anti-TNF treatment [6465]. Genetic study in NLRP1 inflammasome relating to RA pathogenesis was also performed in a Chinese population. Sui et al. examined whether polymorphisms in the locus of NLRP1 gene are linked to susceptibility to RA and showed that NLRP1 gene polymorphism induces up-regulation of NLRP1 gene expression and is a risk factor for RA [66]. These studies clearly indicate that genetic variation by polymorphisms at the loci encoding inflammasomes is strongly associated with RA pathogenesis.
Caspase-1 is a crucial effector molecule in the inflammasome and induces pyroptosis and secretion of interleukin (IL)-1β, one of the critical pro-inflammatory cytokines associated with RA pathogenesis [911]. The role of caspase-1 in RA was evaluated in a caspase-1–/– RA animal model, and the results showed that joint inflammation and cartilage degradation were dramatically reduced in caspase-1–/– mice induced with chronic arthritis [67], indicating that caspase-1 plays a critical role in RA pathogenesis.
Along with the genetic studies of inflammasomes in RA, the role of NLRP1 in RA pathogenesis has been established. Zhang et al. examined the role of 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) in RA pathogenesis and found that inhibition of 11β-HSD1 activity by its specific inhibitor, BVT-2733 significantly ameliorated the symptoms of arthritis including synovial inflammation and joint destruction by decreasing the serum level of IL-1β and suppressing the assembly of NLRP1 inflammasome in collagen-induced arthritic mice compared to controls [68]. Another study also demonstrated the role of NLRP1 in with RA pathogenesis. Li et al. investigated the role of P2X4, a purinergic receptor, in the development of RA in collagen-induced arthritic mice and showed that the inhibition of P2X4 mRNA expression by its antisense oligonucleotide effectively reduced the serum level of IL-1β and also suppressed the activation of NLRP1 inflammasome, leading to reduction of clinical scores in collagen-induced arthritis mice and human RA patients [69].
NLRP3 is another member of the NLR family, and Ippagunta et al. explored the role of NLRP3 inflammasome in RA. They evaluated RA pathogenesis in RA animal models that lack the components of NLRP3 inflammasome. ASC–/– mice were protected from arthritis induction, whereas NLRP3–/– and caspase-1–/– mice were susceptible to arthritis induction [70]. This result was different from the result by Joosten et al. [67], possibly due to the different RA animal models used. Joosten et al. induced arthritis in mice by transferring the serum of K/BxN mice containing the antibodies to recognize glucose-6-phosphate isomerase [71], whereas Ippagunta et al. induced arthritis in mice by injecting collagen (collagen-induced arthritis), suggesting that arthritis induction by different methods might activate different types of immune cells and molecules, possibly resulting in different experimental results. The regulation of NLRP3 inflammasome in RA pathogenesis was investigated. Vande Walle et al. demonstrated that lack of A20, a rheumatoid arthritis susceptibility gene, increased the expression of NLRP3 and pro-IL-1β genes, resulting in induction of NLRP3 inflammasome-mediated caspase-1 activation, pyroptosis, and IL-1β secretion [72]. Moreover, deficiency of NLRP3 significantly suppressed the progression of arthritis and cartilage degradation in A20–/– mice [72], indicating that negative regulation of the NLRP3 inflammasome expression attenuates the pathogenesis of RA. Ruscitti et al. also evaluated the production of IL-1β and activation of NLRP3 inflammasome in the monocytes of RA patients and showed that a significant increase of IL-1β production in the monocytes obtained from RA patients was mediated by the activation of NLRP3 inflammasome [73].
The activation of NLRP3 inflammasome in RA patients was further investigated. Choulaki et al. demonstrated the production of NLRP3 inflammasome components in RA patients and showed that active RA patients had not only higher intracellular levels of NLRP3 inflammasome components including NLRP3, ASC, active caspase-1, and pro-IL-1β, but also increased secretion of IL-1β [74]. Li et al. also examined the activity of NLRP3 inflammasome in arthritic rats. In accordance with previous studies, the activation of NLRP3 inflammasome and the secretion of IL-1β were induced in the synovium of arthritic rats [75]. Inhibition of NLRP3 activity by a clematichinenoside AR, a triterpene saponin extracted from the medicinal plant Clematis manshurica Rupr, suppressed joint inflammation in arthritic rats [75]. Interestingly, a recent study explored the role of NLRP3 in RA pathogenesis using mesenchymal stem cells. Shin et al. administered human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSCs) into collagen-induced arthritis mice and evaluated their therapeutic effect and underlying mechanisms. Systemic administration of hUCB-MSCs dramatically ameliorated the symptoms of arthritis in collagen-induced arthritis mice by suppressing activation of NLRP3 inflammasome in macrophages [76].
Taken together, these results, as described in Fig. 2A strongly indicate that inflammasome activation is a risk factor for the pathogenesis of RA, and selective inhibition of inflammasomes effectively ameliorates RA symptoms and retards RA progression.
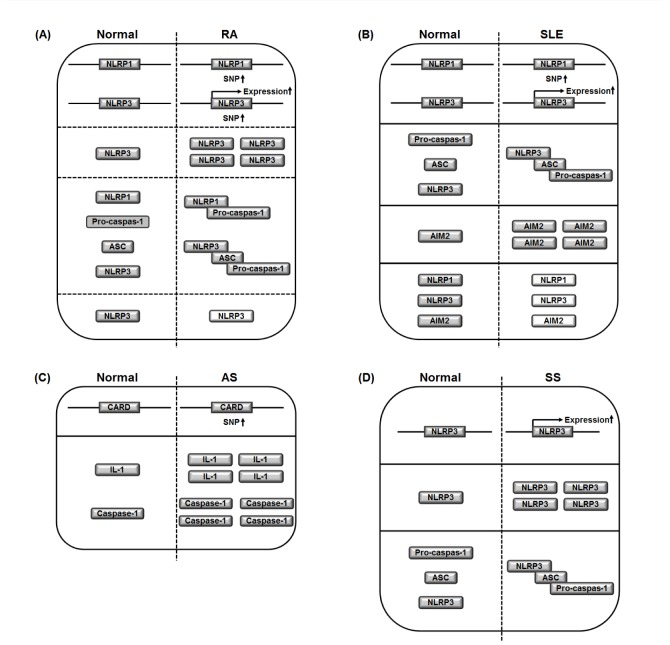 | Fig. 2Graphical summary of the roles of inflammasomes in inflammatory/autoimmune rheumatic diseases.(A) The SNPs and gene expression of NLRP1 and NLRP3, and intracellular NLRP3 level are increased in RA compared to healthy normal control. The activation of NLRP1 and NLRP3 inflammasomes by the assembly with ASC and pro-caspase-1 is induced in RA compared to healthy normal control. In contrast, in a certain condition, NLRP3 knock-out induces RA. A dash line indicates knock-out of NLRP3 gene. (B) The SNPs of NLRP1 and the gene expression of NLRP3 are increased in SLE compared to healthy normal control. The activation of NLRP3 inflammasome by the assembly with ASC and pro-caspase-1 is induced in RA compared to healthy normal control. Intracellular AIM2 level is increased in SLE compared to healthy normal control. In contrast, in a certain condition, intracellular levels of NLRP1, NLRP3, and AIM2 are decreased in SLE compared to healthy normal control. Dash lines indicate the decrease in the intracellular levels of NLRP1, NLRP3, and AIM2. (C) The SNPs of CARD, a critical domain of NLRs are increased in AS compared to healthy normal control. Intracellular levels of IL-1 and caspase-1that are down-stream effector molecules of inflammasomes are increased in AS compared to healthy normal control. (D) The gene expression of NLRP3 and intracellular NLRP3 level are increased in SS compared to healthy normal control. The activation of NLRP3 inflammasome by the assembly with ASC and pro-caspase-1 is induced in RA compared to healthy normal control. SNPs, single nucleotide polymorphisms; NRL, nucleotide-binding oligomerization domain-like receptor; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; AS, ankylosing spondylitis; SS, Sjögren's syndrome.
|
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE), also known as lupus, is a rare inflammatory/autoimmune rheumatic disease that attacks normal healthy tissues in many parts of the body and has a complex pathogenesis. SLE is characterized by various symptoms including inflammatory joints, nephritis, mouth ulcer, swollen lymph nodes, and butterfly-shaped red rash on the face, which is the most typical symptom of SLE [77]. The incidence and prevalence of SLE vary worldwide but is steadily increasing [77]. Although SLE is a rare inflammatory/autoimmune disease, it affects almost all parts of the body, and the symptoms are severe and life threatening. Therefore, many studies have focused on exploring not only the molecular mechanisms of SLE pathogenesis, but also the development of effective therapeutic strategies for SLE.
Active involvement of inflammasomes in SLE pathogenesis has been frequently hypothesized and demonstrated by many research groups. Pontillo et al. examined gene mutations and polymorphisms of inflammasomes and inflammasome effector molecules by SNP analysis in SLE patients and found several SNPs in NLRP1 to be significantly associated with SLE pathogenesis [78].
The role of NLRP3 inflammasome in SLE pathogenesis has been actively explored by many research groups. Shin et al. reported that U1-small nuclear ribonucleoprotein (U1-snRNP), a self nuclear molecule, activated the NLRP3 inflammasome in human monocytes depending on anti-U1-snRNP autoantibodies, which are characteristically found in inflammatory/autoimmune diseases including SLE [79], leading to IL-1β production [80]. This result indicates that self molecules and their autoantibodies can induce inflammatory responses and SLE pathogenesis by activating inflammasomes, such as NLRP3. Neutrophil extracellular traps (NETs) are networks of extracellular fibers primarily consisting of anti-microbial peptides and critical enzymes for host defense [81] and have been suggested to participate in the development of SLE [8283]. Kahlenberg et al. isolated NETs from human SLE patients and demonstrated that NET-mediated activation of NLRP3 inflammasome was significantly enhanced in macrophages derived from SLE patients. Moreover, NETs activated caspase-1, a downstream effector molecule of NLRP3 inflammasome, resulting in secretion of IL-1β and IL-18 in macrophages [84]. These results indicate that increased activation of NLRP3 inflammasome plays a critical role in NET-mediated pathogenesis of SLE.
A pathogenic hallmark of SLE is the autoinflammatory responses against self nuclear antigens, such as circulating double-stranded DNAs (dsDNAs) and anti-dsDNA antibodies, which are found in the sera of SLE patients [8586]. Shin et al. demonstrated that dsDNA and anti-dsDNA autoantibodies induced IL-1β production by activating NLRP3 inflammasome and caspase-1 in human monocytes, leading to production of IL-17 from the IL-1R1+ memory CD4+ T-cells [87]. This plays a crucial role in the pathogenesis of SLE by facilitating production of anti-dsDNA antibodies from B-cells and inducing Th17 cell responses in SLE patients [8889]. The complement component C1q is a risk factor for the development of SLE with its deficiency [90] and was demonstrated to suppress the expression of NLRP3 in human macrophages [91]. Interestingly, the activity of NLRP3 inflammasome was inhibited during C1q-mediated phagocytosis of apoptotic lymphocytes [91], suggesting that activation of NLRP3 inflammasome by C1q absence might contribute to development of SLE. Another study provided direct evidence for the role of NLRP3 inflammasome in the pathogenesis of SLE. Lu et al. examined the role of NLRP3 inflammasome in SLE development using a pristine-induced lupus mouse model and reported that NLRP3 inflammasome was hyperactivated and lupus-like symptoms were more severely developed with higher mortality in the Nlrp3R258W mice that carry a gain-of-function mutation compared to wild type lupus mice [92]. Moreover, a mechanistic study clearly demonstrated that elevated serum levels of anti-dsDNA autoantibodies activated NLRP3 inflammasome and caspase-1 and induced IL-1β secretion in lupus mice [93].
On the other hand, a negative correlation of NLRP3 inflammasome with SLE disease activity was also reported. Yang et al. demonstrated that expression of NLRP3 and NLRP1 inflammasomes was significantly down-regulated in peripheral blood mononuclear cells (PBMCs) derived from patients with SLE compared to those from healthy controls and negatively correlated with SLE disease severity [94]. Lech et al. investigated the role of NLRP3 inflammasome in SLE using mice models lacking the genes of NLRP3 inflammasome components and reported a lack of NLRP3- and ASC-induced lupus-like symptoms in mice [95]. That finding offers further direct evidence that NLRP3 inflammasome plays a pivotal role in the development and progression of SLE. Sester et al. also reported that NLRP3 was absent due to a NLRP3 point mutation, and NLRP3 inflammasome-mediated responses were completely absent in the NZB lupus mice [96].
These results strongly indicate that NLRP3 inflammasome is a risk factor for the development and progression of SLE, while NLRP3 inflammasome negatively correlates with SLE pathogenesis under some conditions.
AIM2 is one of the p200 protein family members to recognize intracellular double-stranded nucleic acids. By recognizing intracellular nucleic acids, AIM2 inflammasome is assembled and activated, leading to activation of caspase-1 and subsequent induction of pyroptosis and IL-1β secretion in macrophages [911]. Formation of AIM2 an inflammasome in response to intracellular double-stranded nucleic acids suggests that an AIM2 inflammasome might be activated in inflammatory/autoimmune diseases, including SLE. Indeed, the serum levels of IL-1β and type I interferons (IFNs) were significantly higher in active SLE patients compared to the normal healthy control [9798]. Zhang et al. further demonstrated that the expression of AIM2 was correlated with severity of disease in SLE patients and a lupus mouse model [99]. Moreover, inhibition of AIM2 expression significantly ameliorated SLE symptoms by suppressing macrophage activation and inflammatory responses in a lupus mouse model [99]. Ding et al. confirmed that mRNA expression level of AIM2 was much higher in spleen, liver, and peripheral blood mononuclear cells of SLE patients than in healthy control individuals [100].
Some other studies, however, have reported that AIM2 inhibition might induce the pathogenesis of SLE. p202 is another member of the p200 protein family and has been reported to be up-regulated in many lupus-prone mouse strains [101102]. p202 has been demonstrated to inhibit AIM2-mediated activation of caspase-1 and to be associated with lupus susceptibility [103104105106]. AIM2 expression in non-lupus prone splenic cells, lupusprone splenic cells, and macrophages expressing high level of p202 was proven to be inversely correlated with p202 expression [107]. In addition, Sester et al. reported that AIM2 inflammasome responses were suppressed due to the high expression of AIM2 antagonist p202 in NZB lupus mice, resulting in low production of IL-1β [96]. These results indicate that AIM2 might be a double-edged sword for SLE pathogenesis.
Lupus nephritis (LN), one of the most serious complications of SLE, is inflammation of the kidneys caused by SLE and the major cause of morbidity and mortality of SLE patients. Many studies have demonstrated the role of inflammasomes in LN during SLE pathogenesis. Gene expression of inflammasome components in LN has been examined. Kahlenberg et al. reported that gene expression of NLRP3 and caspase-1 was significantly up-regulated in LN biopsies [108]. The roles of inflammasomes and their downstream effector molecules, including caspase-1, IL-1β, and IL-18, were also actively explored in LN using various inhibitors and pharmacological compounds. Tsai et al. reported that epigallocatechin-3-gallate, the major bioactive polyphenol, ameliorated symptoms of LN by inhibiting mRNA/protein expression of NLRP3 and the production of caspase-1, IL-1β, and IL-18, which are inflammasome effector molecules in a lupus-prone mice model [109]. Zhao et al. demonstrated that Bay11-7082, a selective inhibitor of the NLRP3 inflammasome, notably decreased serum levels of anti-dsDNA autoantibodies and pro-inflammatory cytokines including IL-1β and TNF-α and ameliorated the symptoms of LN in a lupus mouse model [110]. Zhao et al. also reported that thiadiazolidinone 8 (TDZD-8), a selective inhibitor of glycogen synthase kinase 3β (GSK-3β), inhibited the activation of NLRP3 inflammasome by suppressing caspase-1 activation and IL-1β production in lupus mouse models [111]. Ka et al. further explored the role of NLRP3 inflammasome in LN and reported that citral, a main active ingredient in the herbal medicine Litsea cubeca , significantly inhibited activation of NLRP3 inflammasome and caspase-1 and the secretion of IL-1β. Moreover, it effectively ameliorated LN symptoms in accelerated and severe LN mice [112]. In addition, Li et al. demonstrated that A20, a TNF-induced protein 3 (TNFAIP3) known as a negative modulator of inflammation, markedly suppressed the activation of NLRP3 inflammasome and mitigated LN symptoms in pristine-induced lupus mice [113], and Yuan et al. reported that isoflurane abrogated the formation and activation of renal NLRP3 inflammasome and ameliorated renal dysfunction and injury in MRL/lpr lupus mice [114]. P2X7 receptor (P2X7R), a member of the P2X receptor family, is an ATP-gated ionotropic channel protein mainly expressed on innate immune cells, such as macrophages and dendritic cells [115116]. It has been reported that P2X7R induces the activation of a NLRP3 inflammasome and the secretion of IL-1β by directly interacting with NLRP3 inflammasome scaffold protein in response to extracellular stimulus such as ATP [117118]. Interestingly, Bours et al. explored the role of the P2X7R-inflammasome axis in SLE using lupus mouse models and demonstrated that inhibition of P2X7R showed the beneficial effects in LN in lupus mouse models [119]. Zhao et al. further demonstrated that gene expression of P2X7R-NLRP3 inflammasome signaling molecules, including NLRP3, ASC, and caspase-1, was significantly up-regulated in the kidneys of lupus mice compared to healthy control mice [120]. Moreover, inhibition of P2X7R by its selective inhibitor, brilliant blue G (BBG), suppressed the assembly and activation of the NLRP3 inflammasome and secretion of IL-1β, leading to a significant reduction in nephritis symptoms in lupus mice [120]. In addition to inflammasomes, the role of caspase-1 was also explored in LN. Kahlenberg et al. reported that caspase-1–/– mice were more resistant to lupus and kidney inflammation [121]. The roles of inflammasomes in the pathogenesis of SLE were described in Fig. 2B.
Ankylosing spondylitis and spondyloarthritis
Spondyloarthritis (SpA) and ankylosing spondylitis (AS), the prototype disease of SpA, are types of inflammatory/autoimmune arthritis that primarily affect the axial vertebrae. These diseases cause vertebral fusion which makes the spine less flexible, resulting in a hunched-forward posture and is mainly characterized by severe, chronic pain and discomfort, with significant morbidity and mortality risk [122123]. In spite of the evidence that environmental factors, including gut dysbiosis contribute to developing AS [124], increasing numbers of studies have reported that the disease risk is largely influenced by a genetic factors, and more than 60 genes are associated with the risk of AS [125126127]. Although large-scale studies of the prevalence and incidence of AS are few, the prevalence of AS is generally thought to range from 0.1 to 1.4% worldwide and affects men more often than women [128]. Many studies have examined epidemiological trends in AS. However, these studies have yielded discrepant results due to study design, age, geographic location, genetic susceptibility, disease ascertainment, and ethnicity of patients [128129130131]. In addition, some AS incidence rate reporting has focused only on Europe [131132133134135].
As described earlier, activation of inflammasomes results in the production of the IL-1β from macrophages. Interestingly, the production of IL-1 has been found to be highly induced in AS [136], suggesting the possibility that inflammasomes might be activated and IL-1β production induced in AS. The clinical efficacy of anti-IL-1 monoclonal antibody therapeutics have been examined in AS patients. Tan et al. treated AS patients with Anakinra, an anti-IL-1 monoclonal antibody drug, and reported that Anakinra effectively ameliorated AS symptoms in 67% of the patients [136]. Although this study showed the possibility that inflammasomes could be involved in AS pathogenesis through IL-1β production, it did not provide a direct evidence.
Few studies have reported on the roles of inflammasomes in AS. Therefore, studies to examine the direct role of inflammasomes in AS are needed. Genetic study has established the association between inflammasomes and AS. Kastbom et al. evaluated SNPs in CARD, which is the critical domain of an inflammasome, in 492 AS patients from southern Sweden and reported that a SNP in the CARD8 minor allele was associated with reduced risk of AS [137]. This suggests the possibility that normal inflammasomes containing CARDs might be risk factors for AS pathogenesis.
Activation of caspase-1 is the hallmark of inflammasome activation. Son et al. determined the level of caspase-1 in patients with several types of arthritic diseases including gout, inflammatory arthritis, osteoarthritis, and SpA and reported that caspase-1 level was significantly higher in SpA than in other arthritic diseases [138]. Therefore, caspase-1 could be a biomarker of SpA and helpful in differentiating it from other types of arthritic diseases.
In spite of some studies describing the roles of inflammasomes in AS and SpA (Fig. 2C), studies to provide more direct evidence of the association of inflammasomes in these diseases are needed.
Sjögren's syndrome
Sjögren's syndrome (SS) is a debilitating long-term chronic inflammatory/autoimmune disease in which moisture-generating exocrine gland tissues, such as the tear and salivary glands are primarily targeted by the immune system and is characterized by the development of dry mouth and dry eyes [59139]. The hallmarks of SS are sicca symptoms, but various organ manifestations could also occur [140]. There are two types of SS – primary SS (pSS), which is characterized by loss of salivary and lacrimal fluid, resulting in severe disease manifestations [141], and secondary SS (sSS), which is diagnosed by pSS diagnosis as well as other inflammatory/autoimmune diseases, including RA, SLE, systemic sclerosis, multiple sclerosis, and autoimmune hepatitis and thyroiditis [59142]. The overall prevalence of both pSS and sSS which is more common than pSS is at least 0.4% worldwide with the higher prevalence in Europe [143144], and SS develops more frequently in women than men ranging between 9:1 to 19:1depending on the regions [140].
Although SS is a rare autoimmune/inflammatory disease, many studies have reported the role of inflammasomes in the pathogenesis of SS. The expression level and activity of caspase-1 were examined in SS mouse models, and the results clearly showed that expression and activity of caspase-1 were induced in SS salivary tissues at an early disease state [145146]. This result was confirmed in human SS patients. The expression of inflammasome-related genes, such as P2X7, NLRP3, and caspase-1, was significantly up-regulated in human pSS salivary tissue and correlated with anti-Ro autoantibody presence and focal lymphocytic sialadenitis [147], and the expression of NLRP3 and IL-1β was significantly induced in patients with SS dry eye [148]. The production of both IL-1 and IL-18 resulting from inflammasome activation was highly induced in human SS patients as well as SS mouse models [145147149150151152153]. These studies indicate that the inflammasome is actively involved in the onset and development of SS.
As discussed earlier, P2X7R directly interacts with and activates the NLRP3 inflammasome, resulting in induction of IL-1β secretion, and the role of the inflammasome in the P2X7R-inflammasome axis was also explored in SS using an animal model. P2X7R-deficient mice were protected from inflammation of a salivary gland, and activation of P2X7R by local delivery of P2X7R agonist in mice induced severe inflammation in a salivary gland [154], indicating that the activation of P2X7R could induce SS-like symptoms in mouse models by activating NLRP3 inflammasome. Another study reported the role of the P2X7R-inflammasome axis in human SS patients. They showed that expression of the genes in the P2X7R-inflammasome axis, including P2X7R, NLRP3, caspase-1, and IL-18, was significantly induced in the salivary gland specimens of SS patients [147].
Taken together, these studies, as described in Fig. 2D strongly suggest that the activation of inflammasome, especially NLRP3 inflammasome, in salivary gland tissue might be a critical event for the pathogenesis and progress of SS.
Go to :
CONCLUSION AND PERSPECTIVES
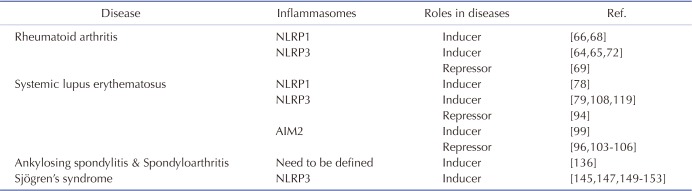
Inflammation is an innate immune response mediated mainly by macrophages in order to protect the body from pathogen invasion. One hallmark of macrophage-mediated inflammatory responses is activation of inflammasomes. Inflammasomes are intracellular protein complexes that can activate caspase-1, resulting in the maturation and secretion of pro-inflammatory cytokines such as IL-1β and IL-18 and induction of pyroptosis. They include NLR family inflammasomes, including NLRP1, NLRP3, and NLRC4 as well as non-NLR family inflammasomes, such as AIM2. Although activation of inflammasomes during the inflammatory response is a host defense mechanism, these inflammasomes play critical roles in chronic inflammatory responses that are regarded as the major cause of inflammatory/autoimmune rheumatic diseases. Several studies have demonstrated that inflammasomes and downstream effector molecules including caspase-1, IL-1β, and IL-18 are highly expressed and activated in inflammatory/autoimmune rheumatic diseases, and that the activation of inflammasomes is a risk factor for the development and progression of these diseases. On the other hand, several studies have also claimed that some types of inflammasomes, such as NLRP3 and AIM2, negatively correlate with the pathogenesis of these diseases under certain conditions. The roles of inflammasomes in each inflammatory/autoimmune disease are summarized in Table 2.
In spite of a number of successful studies reporting the roles of inflammsomes in the inflammatory/autoimmune rheumatic diseases, no study demonstrating the roles of non-canonical inflammasomes in these diseases has been reported. In addition, no study investigating the functional crosstalk between canonical and non-canonical inflammasomes as well as between different types of canonical inflammasomes during the pathogenesis of these diseases has been reported. Therefore, these studies need to be further investigated to explain how they cooperate and make synergistic or antagonistic effects during the pathogenesis of these diseases. Moreover, extensive studies on the useful strategies to selectively target and regulate the activation of inflammasomes and their pathways during inflammatory responses, such as inflammasome-specific small interfering RNAs, a recently developed inflammasome-targeting intracellular antibody technology [155], and the agents to inhibit the inflammasome assembly are highly required for developing effective drugs that could be used for these diseases. Given strong evidence of the critical role of inflammasomes in macrophage-mediated inflammatory responses and in inflammatory/autoimmune rheumatic diseases, targeting inflammasomes represents a novel and promising strategy for the prevention and treatment of inflammatory/autoimmune rheumatic diseases.
Go to :
 XML Download
XML Download