

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Peripheral arterial disease is a common complication in patients with diabetes [1]. Hyperglycemia causes vascular damage with several underlying mechanisms. Among them, high glucose levels in the blood may contribute to endothelial dysfunction by mediating abnormal endothelial permeability or an imbalance between endothelium-derived relaxing and contracting factors [2345678]. Several previous studies also demonstrated that prolonged exposure to high glucose may affect endothelium-dependent vasodilation by stimulating the production of reactive oxygen species (ROS) [9101112] or voltage-gated K+ channel function in coronary vascular smooth muscle cells [13]. The majority of previous studies demonstrated effects of hyperglycemia on vascular functions with long-term exposure; however, a few studies have been conducted to determine the effects of high glucose with short-term exposure. The results of those studies varied with the different experimental conditions [141516]. The incubation of high glucose (20 mM) for 2 h induced impairment of bradykinin-induced relaxation in human subcutaneous arteries whereas induced opposite result in human mesenteric arteries (MAs) [15]. Also, exposure of high glucose (44 mM) for 3h produced the attenuation of phenylephrine (PhE)-induced contraction in rat aortic rings [14].
Free fatty acids (FFAs) regulate vascular functions, and circulating FFA levels in blood serum are often associated with hyperglycemia. Some studies have demonstrated that excessive levels FFAs may cause vascular dysfunction by enhancing oxidative stress or reducing nitric oxide (NO) production in endothelial cells [17181920]. A previous study has suggested that exposure to FFAs at both high and physiological concentrations impaired endothelium-dependent relaxation (EDR) in MA [21]. However, results from another study showed that FFAs did not affect endothelial function or ROS generation in the vessels [20].
Conventional in vitro studies of arterial contractility is conducted in physiological salt solutions with glucose only for the metabolic substrates. Considering the importance of vascular functions in the pathophysiology of metabolic diseases, it is requested to have a comprehensive understanding of systemic arterial contractility under physiological conditions. On these backgrounds, we investigate the effects of short-term incubation with high glucose with or without other metabolic substrates on α-adrenergic agonist-induced contraction in rat mesenteric arteries (MAs) and deep femoral arteries (DFAs).
METHODS
Preparation of arterial rings
Experiments were performed with the approval of the Institutional Animal Care and Use Committee in Chung-Ang University (approval no. 2015-00012). Adult male Sprague-Dawley rats (240~320 g) were anesthetized by intraperitoneal injection of pentobarbital sodium (60~100 mg/kg). Full anesthesia was confirmed by the absence of limb withdrawal using toe pinching. The intestine and proximal hind limbs were removed quickly and placed into normal Tyrode (NT) solution at 4℃. The NT solution contained the following (in mM): NaCl 140, KCl 5.4, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid 10, NaH2PO4 0.33, glucose 10, CaCl2 1.8, and MgCl2 1. The NT solution was adjusted to pH 7.4 with NaOH. Arteries were dissected from fat and connective tissue in NT solution. The second and third branches of MAs and DFAs were isolated quickly (inner diameter: 200~300 µm) and cut into segments 2.5~3 mm in length.
Tension measurement protocol
Endothelium-intact artery rings were mounted on two 25 µm tungsten wires in a dual-wire myograph (620 M; DMT, Aarhus, Denmark). Each myograph chamber was filled with 5 mL of physiological salt solution (PSS). An arterial ring segment was stabilized in PSS aerated with 21% O2,/5% CO2 (the remainder N2) at 37℃. The PSS solution consisted of the following (in mM): NaCl 118, KCl 4, MgSO4 1, NaHCO3 24, NaH2PO4 0.44, glucose 5.6, and CaCl2 1.8.
For each vessel, 80 mM KCl-PSS was used to obtain initial maximal contractility. To compare between groups, the contractile responses induced by different concentrations of phenylephrine (PhE) were normalized to the 80 mM KCl-induced maximum constriction in each vessel. In addition, EDR was measured by the response to 10 µM acetylcholine (ACh) with rings pre-contracted with 10 µM PhE. The contractile response induced by PhE or KCl was assessed in the presence or absence of high glucose (23 mM), or with metabolic substrates.
Solutions and chemicals
Metabolic substrates were added to the PSS solution. The metabolic substrate compositions were modified based on analyses of the human's blood sample [22]. The NF solution consisted of the following (in mM): 96 mL PSS, oleic acid 200 µM, palmitic acid 100 µM, linoleic acid 100 µM, sodium L-lactate 1 mM, sodium pyruvate 100 µM, and carnitine hydrochloride 50 µM. All fatty acids were prepared at physiological concentrations. Palmitic acid, linoleic acid, and oleic acid were dissolved in 0.1 N NaOH. All drugs and chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysis
Data are shown as means±SEM. The response to PhE is expressed as the percent of the second 80 mM KCl-PSS-induced maximum constriction. Half-maximal effective concentrations (EC50 levels) were calculated by non-linear regression analyses of the concentration response curves generated with OriginPro 8.0 software (OriginLab, Northampton, MA, USA). Statistical analysis was performed with the unpaired Student's t-test. Statistical significance is reported as p<0.05.
RESULTS
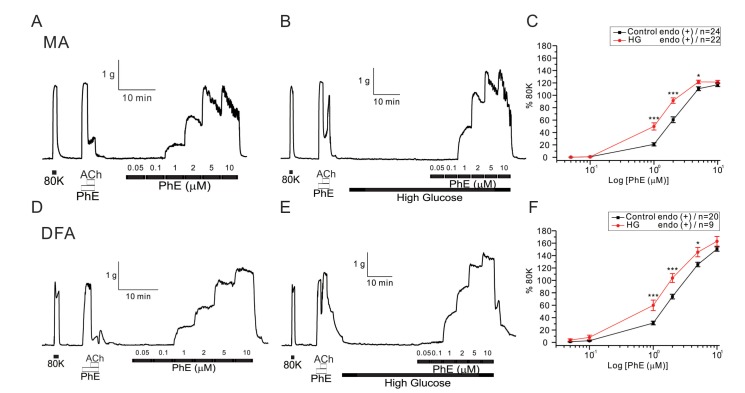
After confirming 80 mM KCl-induced (80K contraction) and 10 µM PhE-induced contraction, endothelium-dependent ACh-induced vasorelaxation was tested in each vessel. The tested vessels were either incubated with 23 mM glucose-containing or control PSS, and then the concentration-dependent contractile responses to PhE were analyzed. Incremental application of PhE (0.05~10 µM) induced stepwise increase of arterial tones in both MAs and DFAs (Fig. 1). Noticeable contraction was observed from 1 µM PhE. In general, the PhE- contractions at 2 µM and above were more persistent in DFAs than MAs; MAs showed partial relaxation following peak contractions. Thus, for the comparison between MAs and DFAs, the peak amplitudes of PhE- contraction were analyzed after normalization to 80 K contraction in each vessel. After 30 min incubation in the presence of high glucose (23 mM, HG), the amplitudes of PhE-contraction became higher in both MAs and DFAs (Fig. 1). The EC50 values of PhE-contraction in MAs were 1.96±0.37 and 1.22±0.25 µM for control and HG, respectively (Fig. 1A), and 2.41±0.14 and 1.61±0.34 µM for control and HG in DFAs (Fig. 1B), respectively.
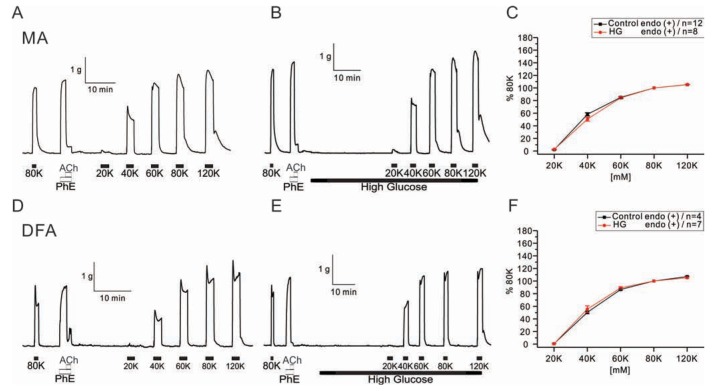
To determine the effect of HG on the purely membrane depolarization-dependent contraction, cumulative doses of KCl were applied to both arteries (Fig. 2). Contractile responses to KCl were unaffected in the presence of HG (Fig. 2C, F).
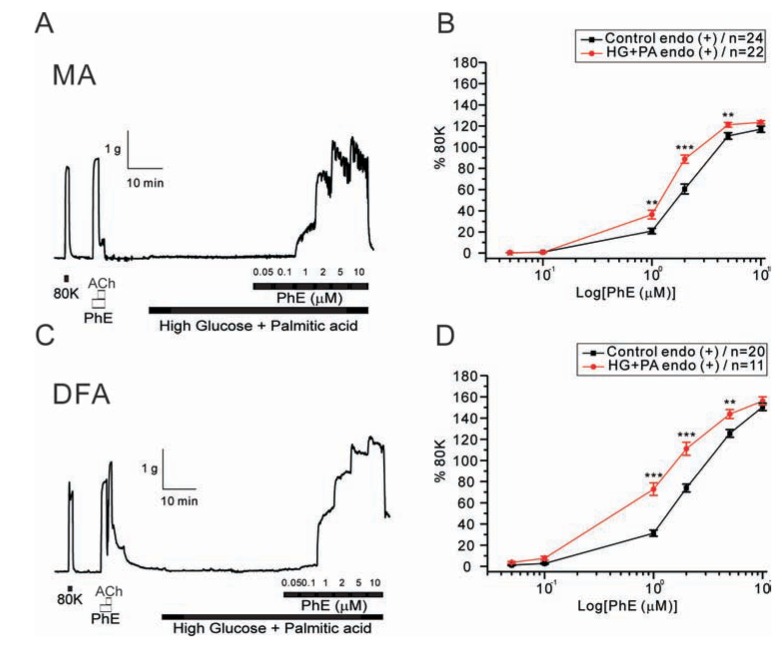
Next, we tested whether combined application of HG and saturated fatty acid differently affects the PhE- contractions. When co-incubated with palmitic acid (100 µM), the enhancement of PhE-contraction by HG was similarly observed in MAs and DFAs (Fig. 3). The EC50 values were: control MAs, 1.96±0.27 µM; high glucose MAs, 1.41±0.33 µM; control DFAs, 2.41±0.19 µM; high glucose DFAs, 1.20±0.05 µM. It has to be noted that the concentrations-dependent PhE- contraction data for the control condition were same ones in Fig. 1 and Fig. 3.
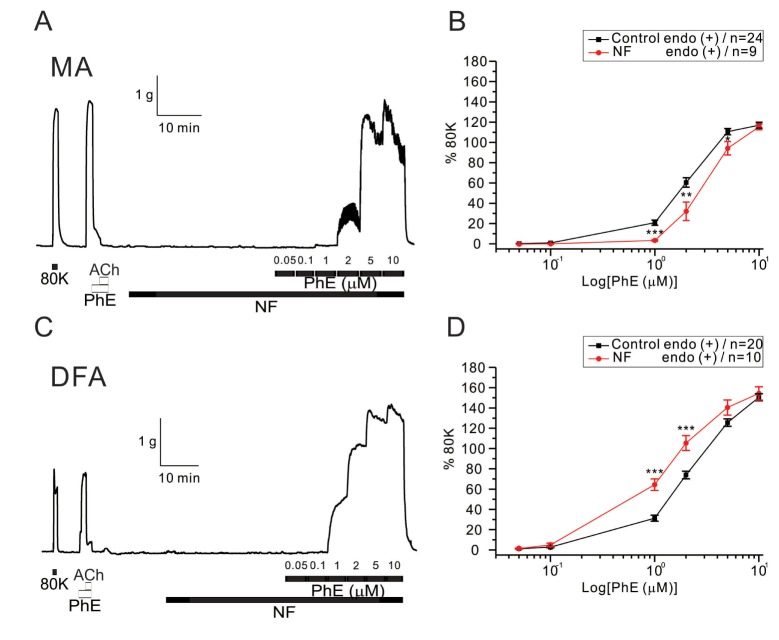
Then we tested the effects of NF solution on PhE-contractions. A significant reduction of PhE (1~5 µM)-induced contraction was observed when incubating MA rings with NF solution for 30 min (Fig. 4A, B). EC50 values were: control, 1.96±0.31 µM; NF, 3.08±0.38 µM. Interestingly, however, the incubation of DFA rings with NF solution for 30 min produced an increased sensitivity to PhE-induced contraction (Fig. 4). The NF solution caused a significant leftward shift of the PhE (1 and 2 µM) concentration-response curve (Fig. 4C, D). EC50 values were: control, 2.41±0.14 µM; NF, 1.36±0.22 µM.
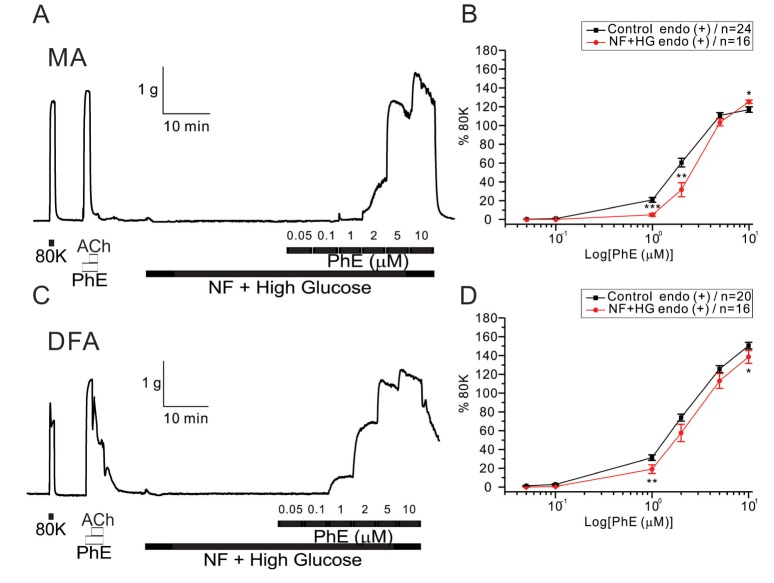
The co-incubation with HG in NF solutions did not increase but slightly decreased the sensitivity to PhE- contraction in MAs (Fig. 5A, B). EC50 values were 1.96±0.28 and 3.07±0.27 µM for control and HG with NF in MAs, respectively (Fig. 5B). More interestingly, the enhanced PhE- contraction of DFAs under NF solution became reversed to inhibition smaller than the control level. Significant rightward shifts of the concentration-response curves to PhE (1 and 2 µM in MAs, 1 and 10 µM in DFAs) were observed in the presence of the combined high glucose and NF solutions (Fig. 5B, D). EC50 value was 2.99±0.18 µM for HG/NF in DFAs (Fig. 5D), respectively.
DISCUSSION
The results of the present study showed that only 30 min of incubation in HG enhances the sensitivity to PhE-induced contraction in rat MAs and DFAs. More interesting finding was that the incubation in NF solution induces opposite responses, decrease and increase of PhE-contractions in MAs and DFAs, respectively. Furthermore, the pro-contractile effect of HG alone converts to an attenuation of PhE-contraction in NF solution, especially in DFA.
The effects of HG on PhE-induced contraction vary depending on the experimental conditions [1415162324]. Although long term exposure to high glucose inhibits EDR in a majority of studies [252627], several studies show conflicting results with regard to the effects of short-term exposure to high glucose [14151628]. A previous study using diabetic rats showed decreased sensitivity to PhE-induced contraction in blood vessels [24] whereas another study demonstrated the opposite result [29]. A more recent study also reported that PhE-induced contraction in aortic rings was attenuated by acute exposure to high glucose (44 mM) for 3 h [14].
In the conventional PSS solution, the acute exposure to HG consistently enhanced the PhE-contraction in both MAs and DFAs (Fig. 1). Because the high K+-induced contractions were not affected by HG, the procontractile effects appears to be more specifically associated with the a-adrenergic signaling pathways or might be mediated by K+ channel inhibition [13]. The absence of any change in the high-K+ contraction also suggests that the endothelial function is not acutely changed by HG for 30 min. This supposition is supported by a previous finding that the relaxation produced by ACh was induced by a 6 but not 2 h incubation in high glucose (20 mM) in human mesenteric and subcutaneous arteries [15].
However, we could not exclude the effects of acute HG on endothelial functions. Several studies demonstrated that impairment of EDR augmented PhE-induced contraction following prolonged exposure to high glucose [5]. In addition, a high concentration of glucose induced endothelial dysfunction as shown by decreased ACh-induced NO-mediated relaxation [7830] and increased production of ROS in cultured endothelial cells [718]. NADPH oxidase (NOX) is one of the most important sources of ROS [31]. Several studies have indicated that high glucose activates NOX and induces ROS production in endothelial and vascular smooth muscle cells via a protein kinase C-dependent pathway [3233], and that ROS interact with endothelial NO to reduce NO bioactivity [34]. Therefore, a decrease of EDR by short-term exposure to high glucose could affect increased sensitivity to PhE-induced contraction in MAs and DFAs.
The glucose concentration we applied in the present study was similar to postprandial glucose levels in Type II diabetic patients [35]. Initially, it was suggested that the augmentation of PhE-contraction by the acute HG suggested that the total peripheral resistance under tonic sympathetic tone might induce an increase of blood pressure after some heavy meals. However, the differential effects of HG on PhE-contractions in NF solution imply that the actual influence of glucose uptake on the resistance of circulation system requires careful interpretation.
In the present study, the effects of NF solution were demonstrated in rat blood vessels (Fig. 4). In rat MAs and DFAs, the application of NF solutions showed inconsistent effects on PhE-induced contraction. The mechanism underlying this inconsistency is unclear. However, we cautiously assume that the specific composition of the NF solution may reduce sensitivity to PhE-induced contraction in MAs. At first, we assumed that saturated fatty acids such as palmitic acid might be responsible for the differential responses in NF. However, it was found that the sensitivity to PhE-induced contraction was consistently augmented by co-incubation with palmitic acid and high glucose in both arteries (Fig. 3). Thus, other components of NF, or more complicated effects of the combined components seem to be responsible for the intriguing influence of NF solution.
Previous studies showed that individual free fatty acids have similar effects in MAs and DFAs, and endothelial cell dysfunction was induced with high and physiological concentrations of both saturated and unsaturated fatty acids [2136]. Further investigation is needed to identify these inconsistent effects of NF solutions using various arteries and solution compositions.
In conclusion, the results of the current study demonstrated that short-term exposure to a high glucose solution augmented sensitivity to PhE-induced contraction in rat MAs and DFAs. In addition, this is the first study to examine the effects of NF solution alone and with high glucose. These findings provide preliminary evidence for identifying the action mechanism of metabolic substrates and high glucose on vascular reactivity.
 XML Download
XML Download