

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Aging results in the structural and functional degradation of the cardiac muscles accompanied by reduced contractility and muscle mass. Cell loss or reactive hypertrophy in aged heart results in functional degeneration, leading to eventual defects in heart function [1]. Changes in muscle cells during the aging process is mainly caused by metabolic changes from qualitative and quantitative changes in the mitochondria, fluctuations in the levels of oxidative stress factors in cells, changes in the Ca2+ signaling structure, and the transformation of type II muscle fibers to type I muscle fibers [2]. Since 95% of the energy required for contraction of the cardiac muscle are supplied by mitochondrial respiration, these metabolic changes have a significant impact on heart dysfunction and can lead to the onset of heart failure [3]. Thus, enhancing mitochondrial biogenesis of the heart is crucial for maintaining heart function and preventing cardiac diseases.
Along with aging, obesity is another key risk factor of cardiovascular diseases. Obese people have increased levels of plasma free fatty acids, with consequent activation of endoplasmic reticulum (ER) stress and a chronic inflammatory response with negative effects on the condition of the heart [4]. ER stress is strongly associated with various cardiovascular diseases, including heart failure, arteriosclerosis, and ischemic heart diseases [567]. A recent study showed that increased ER stress resulted in a 2.4-fold reduction in
mitochondrial biogenesis [8]. It is well known that the signaling pathway in ER stress mediated by three major ER stress sensors which are PKR-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). PERK of them is a key ER stress sensor and molecular mediator of the ER-mitochondria contact sites, required to regulate inter-organellar cross-talk in ROS-induced cell death, a role which is not shared with other ER stress sensors, like IRE1 [9]. Therefore, PERK and pro-apoptotic C/EBP homologous protein (CHOP), a downstream of PERK, are key factors to understand an association between ER stress and mitochondria malfunction caused by obesity, but these are not clearly understood.
One of the most effective methods to stimulate mitochondrial biogenesis within the muscle tissue is through an exercise program. More specifically, high-intensity or long-term exercise training have been shown to increase the number and quality of mitochondria [1011]. Previous studies have shown that endurance exercise training in elderly subjects increased the VO2 peak [121314], mitochondrial quantity [15], oxidase activity [1214], muscular protein synthesis rate [1617], mitochondrial protein gene transcripts [13], and the copy number of mitochondrial DNA [18]. Furthermore, physical exercise has shown to reduce the levels of oxides [19] and to protect muscle cells by increasing the activity of antioxidant enzymes [202123] that is associated with peroxisome proliferator-activated receptors β/δ (PPARδ) [242526]. PPARδ is an essential mediator to reduce ER stress in aortas of obese mice [27], and is required for exercise to attenuate ER stress [28], therefore, these findings led us to postulate that the PPARδ induced by exercise reduces ER stress in cardiac muscle caused by obesity-induced ROS.
The aim of the present study was to determine the effects of PPARδ on the mitochondrial enzymes and PERK/pro-apoptotic C/EBP homologous protein (CHOP) pathway, a key ER stress pathway, in the cardiac muscle of high fat diet-induced obese middle-aged rats that were perform to high-intensity intermittent training using ladders and weights. The results of this study expect to provide key evidence to support the benefits of exercise training for maintaining cardiovascular health and preventing heart diseases through a mechanism of enhancing the obesity-incurred reduction of mitochondrial biogenesis.
METHODS
Subjects
After a 1-week adaptation period, 40 male, middle-aged (~50 weeks old) Sprague-Dawley rats were purchased and induced to become obese with 6 weeks of a high-fat diet (30%, 50%, and 20% calories from carbohydrate, fat, and protein, respectively). After obesity induction, the rats were randomly divided into four groups: the normal chow diet group (a high-protein diet consisting of 64.5%, 11.8%, and 23.5% calories from carbohydrate, fat, and protein, respectively; 3.2 kcal/g); the high-fat diet group (5.1 kcal/g); the chow+exercise group; and the high-fat diet+exercise group. The treatment regime continued for a total of 8 weeks. The exercising groups underwent high-intensity intermittent training using a ladder-climbing exercise 3 days/week. The chow diet groups were switched from the initial high-fat diet to the chow diet to reduce the total caloric intake and fat consumption.
The rats of all groups received concurrent supplements of vitamins (22 g/kg Teklad vitamins mix no. 40077), minerals (51 g/kg Teklad mineral mix no. 170915), methionine (5 g/kg, Teklad Premier no. 10850), and choline chloride (1.3 g/kg) [29]. Drinking water was provided ad libitum in the form of secondary ion-exchange water. All rats were individually bred during the breeding period, and the day-night cycle was 12 h. Temperature and relative humidity were maintained around 24±1℃ and 60%, respectively. The study design was approved by the Animal Research Ethics Committee of Keimyung University School of Medicine (KM-2012-30R).
Ladder-climbing exercise
For adaptation, the rats were trained on the ladder-climbing exercise for the first week, using a 1-m ladder positioned at a 75° incline without adding a weight to the tail. After this adaptation period, weights of 30~50% body mass were added to the tails, and the weight or number of repetitions was steadily increased during the experimental period. For the first set, a weight of 70% body weight was added from the 2nd week until the end. An 80% weight was added for the 2nd and 3rd sets, a 90% weight was added for the 4th and 5th sets, and a 100% weight was added for 6th to 8th sets. This protocol for rodent's resistance exercise was followed or modified from Kraemer et al.'s protocol [30]. There were 8 repetitions per set, with a 2 min resting period between sets. The frequency of the ladder-climbing exercise was 3 days per week [3132].
Tissue extraction and analysis
After 8 weeks of the treatment, the rats were given a 48 h resting period to eliminate any last-bout exercise effect. The rats were then fasted for 12 h, and anesthetized using Zoletil 50 (10 mg/kg body weight, Virbac Korea) and Rumpun 2% (0.04 ml/kg, Bayer Korea). First, the abdominal cavity was opened and blood was extracted from the abdominal arteries. The heart was then extracted after completely removing the blood via perfusion using phosphate-buffered saline. The weight was measured once after removing the water contents, and then the heart was snapfrozen and stored at −80℃ until protein analysis. The extracted heart was homogenized using ice-cold buffer [250 mM sucrose, 10 mM HEPES/1 mM ethylenediaminetetraacetic acid (EDTA, pH 7.4), 1 mM Pefabloc (Roche), 1 mM EDTA, 1 mM NaF, 1 µg/ml aprotinin, 1 µg/ml leupeptin, 1 µg/ml pepstatin, 0.1 mM bpV (phen), 2 mg/ml glycerophosphate]. The homogenized sample was subjected to three freeze/thaw cycles and centrifuged (700 g, 10 min). The supernatant was collected, and the protein level was quantified through the Bradford assay [33]. The prepared sample was dissolved in Laemmli buffer, loaded onto a sodium dodecyl sulfate-polyacrylamide gel, electrophoresed, and then transferred to a nitrocellulose membrane. The membrane was blocked for 60 min using 5% non-fat dry milk and Tris-buffered saline+0.1% Tween 10 (TBST; pH 7.5), washed with TBST, and incubated with the following primary antibodies overnight at 4℃: phospho-protein kinase RNA-like endoplasmic reticulum kinase (PERK, Cell Signaling Technology, Beverly, MA, USA), PERK (Cell Signaling Technology, Beverly, MA, USA), peroxisome proliferator-activated receptor (PPAR) β/δ (Santa Cruz Biotechnology, Santa Cruz, CA, USA), C/EBP homologous protein (CHOP, Cell Signaling Technology, Beverly, MA, USA), 78-kDa glucose-regulated protein (GRP78, Santa Cruz Biotechnology, Santa Cruz, CA, USA), succinate dehydrogenase (Santa Cruz Biotechnology, Santa Cruz, CA, USA), cytochrome C (Santa Cruz Biotechnology, Santa Cruz, CA, USA), peroxisome proliferator-activated receptor-coactivator 1α (PGC-1α), and β-actin (Calbiochem, Germany). After another wash with TBST, the samples were treated with the secondary antibody (anti-mouse or anti-rabbit, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 60 min. The bands were visualized using ECL (Genekhan Scientific, St. Louis, MO, USA), and the relative intensity of the bands was assessed using SigmaGel (Jandel Scientific Corp., Erkrath, Germany).
RESULTS
Body weight and visceral fat weight
Feeding of the high-fat diet for 8 weeks resulted in a significant increase of both body weight and visceral fat weight for the middle-aged rats (p<0.05). Concurrent ladder climbing exercise with the high-fat diet did not result in a significant reduction of body weight, but did lead to a significant reduction of visceral fat weight (p<0.05). The groups that were switched to the chow diet for 8 weeks had significantly reduced body and visceral fat weights (p<0.05) compared to those of the high-fat diet group. The group on the chow diet with exercise showed a reduced body weight and visceral fat weight compared to the chow diet-only group, but the difference was not statistically significant (Fig. 1).
Mitochondrial biogenesis enzymes
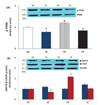
The protein expression levels of cytochrome C and succinate dehydrogenase were measured to evaluate the effects of diet and/or exercise on mitochondrial biogenesis within the cardiac muscle. Eight weeks of ladder-climbing exercise significantly increased the mitochondrial enzyme levels (p<0.05). As shown in Fig. 2, the mitochondrial enzyme levels were particularly significantly higher in the chow diet+exercise group compared to those in the other three groups (p<0.05).
PPARδ and PGC-1α protein levels
To identify the potential mechanism by which the 8-week ladder-climbing exercise promoted mitochondrial biogenesis within the cardiac muscle of middle-aged rats, the protein levels of PPARδ and PGC-1α were measured. The level of PPARδ in the cardiac muscle was significantly increased in the high-fat diet+exercise group, but there were no significant changes in the other three groups. The level of PGC-1α did not differ among any of the treatment groups (Fig. 3).
ER stress-related biomarkers
To assess the effects of diet and/or exercise on ER stress within the cardiac muscle, the levels of phospho (p)-PERK, PERK, and CHOP proteins (markers of myocardial damage) and GRP78 protein (marker of myocardial protection) were measured. Maintenance on the high-fat diet resulted in a significant increase in p-PERK and CHOP protein levels (p<0.05), whereas the ladder-climbing exercise significantly reduced these levels down to values comparable to those of the chow diet group (p<0.05). More specifically, the CHOP protein level in the chow diet+exercise group was significantly lower than that of the chow diet-only group (p<0.05), representing the lowest level of CHOP protein overall. However, there was no significant difference in the GRP78 protein level among the groups (Fig. 4).
DISCUSSION
In this study, the effects of intermittent ladder-climbing exercise training on mitochondrial biogenesis within the cardiac muscle of middle-aged rats were assessed by exploring the roles of a biogenesis-promoting mechanism (PGC-1α) and biogenesis-inhibiting mechanism (ER stress). Recent studies have demonstrated that ER stress is a key factor contributing to the quantitative and functional degradation of the cardiac skeletal muscle during the aging process [34]. The ER is one of the organelles found in all eukaryotic cells, composed of a web-shape mixture of tubules, vesicles, and cisternae. The ER produces proteins that are distributed to various compartments of the cell. When an immature protein enters the ER that cannot be handled by the organelle owing to changes in the physiological or pathological environment or when the calcium within the ER is depleted, the ER cannot function properly, reaching a state of ER stress [353637]. ER stress is strongly associated with various cardiovascular diseases such as heart failure, cardiotoxicity, arteriosclerosis, ischemia, and heart diseases [5], and many studies have been performed to investigate the potential of exercise training for reversing these changes in ER stress to prevent or treat these conditions. Murlasits et al. [38] placed 16-week-old rats on a 60 min running exercise routine at an intensity of VO2max >70% for 5 days, and found no effect of this short-term endurance training on the expression of ER stress-related proteins in the cardiac muscle, such as GRP78, GRP94, or calreticulin. However, Gonzalez et al. [39] reported that endurance training for 3 months resulted in significantly increased expression of GRP78 within the soleus and extensor digitorum longus muscle. GRP78 and GRP94 are ER chaperones that protect myocardial cells against damage (i.e., cell death) induced by myocardial ischemia when activated [4041]. However, the effect of exercise on ER stress is still unclear, requiring continuous and detailed studies to determine the underlying mechanisms.
High-intensity or long-term exercise training increases the number and quality of mitochondria by effectively stimulating mitochondrial biogenesis [10]. Although the reasons for this adaptation mechanism are still unclear, recent microarray analyses have suggested that this phenomenon is caused by the exercise-induced promotion of the transcription of mitochondrial proteins to consequently increase the expression of PGC-1α [424344454647]. However, in our study, in spite of no significant change in the level of myocardial PGC-1α after 8 weeks of a ladder-climbing exercise, the PPARδ level increased in the exercise groups, with a significant increase in the high-fat diet+exercise group compared to that in the other groups. PPARs are nuclear receptors that use free fatty acids as ligands for their activation [48], and show tissue-specific expression patterns. PPARα and PPARγ are specifically expressed in the liver and fat tissue, respectively, and PPARδ expression is extremely high in tissues with relatively high metabolic activity, including the muscles, liver, and fat tissues [49]. Previous studies [505152] have demonstrated that PPARδ could play a role in increasing running endurance and the oxidative metabolic activity of the skeletal muscle in rats. Wang et al. [52] established transgenic mice with overexpression of PPARδ in the skeletal muscle, which had increased levels of mitochondrial enzymes without a corresponding increase in the PGC-1α mRNA level. Nuclear respiratory factor-1 (NRF-1) is a key transcription factor for nuclear-encoded subunits of the four mitochondrial respiratory chain complexes and ATP synthase [5354], and PPARδ has been identified as a transcription factor for NRF-1 [54]. PPARδ overexpression has been shown to increase SUO and Cyt C as well as NADH and ATP synthase prior to the increase in PGC-1α expression [55]. Therefore, we suggest that intermittent ladderclimbing exercise training is supposed to increase mitochondrial biogenesis through the expression of PPARδ without a significant change of PGC-1α in aged myocardial tissue.
The results of the present study showed that the higher levels of the myocardial damage-associated proteins p-PERK and CHOP in the cardiac muscle of high fat diet-induced obese middle-aged rats could be significantly reduced by 8 weeks of intermittent ladder-climbing exercise training. High fat diet increases the production of mitochondrial ROS which is clearly associated with insulin resistance [56575859]. Reactive oxygen species (ROS) has been identified as crucial regulators of ER function and unfolded protein response activation (UPR), ER stress occur concurrently with an increased ROS production [60]. ROS modulates the cross-talk between ER and mitochondria during apoptosis caused by ER stress [6162]. Previous study has shown that PERK, a key ER stress sensor of the UPR, is an essential component of mitochondria-associated ER membrane (MAMs) [9]. MAMs were established a physical and functional connection between ER and mitochondria [63]. Thus, an increased PERK phosphorylation in cardiac muscle due to chronic high fat diet involved in an increase ROS and ER stress. Superoxide dismutase2 (SOD2) [24] and catalase [25] promoter has PPAR response element, Fan et al. [26] showed that PPARδ activation increases Glutathione Peroxidase 1 (GPx1), SOD3 and catalase gene in skeletal muscle. It is well known that exercise training decreases ROS and increases PPARδ as well as antioxidants in cardiac muscle (REF). In present study, therefore, an increased PPARδ in cardiac muscle by intermittent ladder-climbing exercise training of rat resulted in a decrease PERK/CHOP pathway that is associate with ROS [9].
This reduction was even greater in the group with lower caloric intake owing to switching from the high-fat diet to chow diet with concurrent exercise training. These results indicate that intermittent ladder-climbing exercise training combined with a reduced caloric intake can attenuate the levels of ER stress-related proteins that would induce myocardial damage. However, there was no influence of exercise or diet on GRP78 levels, which is a protein related to myocardial protection. Previous studies reported that acute exercise significantly increased the GRP78 level in the liver, but the effects on other tissues were not consistent [64]. Similarly, in our study, there was no change in GRP78 expression in the cardiac tissue, although changes are possible in other tissues.
The results of the present study suggest that changes in ER stress-related factors caused by intermittent high-intensity training in the context of aging do not play a substantial role in the activation of mitochondrial biogenesis, or that these changes are mutually independent of each other. However, considering previous findings suggesting that ER stress can lead to the suppression of mitochondrial biogenesis [65], it is crucial to clearly identify this potential association through additional analyses of reactive oxygen species induction and defense-related factors [66]. Moreover, the exercise routine used in this study, i.e., adding weights to the tail with ladder climbing, is considered an intermittent high-intensity type of training, and it is possible that persistence in the stimulation from exercise was not sufficient to alter the PGC-1α level in the cardiac muscle. Finally, the use of middle-aged rats rather than young, healthy rats should also be taken into consideration when interpreting these results. Therefore, along with age- and tissue-specific changes, associations between ER stress, PPARδ, and PGC-1α should be identified through future studies.
One particularly interesting finding of this study was that switching the high fat-induced obese rats to a chow diet for 8 weeks caused a significant decrease in body weight and visceral fat weight, but did not affect myocardial mitochondrial biogenesis. Civitarese et al. [67] analyzed the effect of diet (i.e., reduced caloric intake) or endurance training over 6 months on the mitochondrial biogenesis of the skeletal muscle in 36 obese human subjects, and claimed that reduced caloric intake improved mitochondrial function. However, conflicting results have emerged from animal studies evaluating the effects of reduced caloric intake on mitochondrial biogenesis of the skeletal muscle [68697071]. Furthermore, even among studies on humans, there are many reports showing no increase in the activation of oxidative enzymes in obese subjects after weight loss [7273].
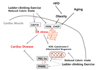
The results indicated that 8 weeks of a ladder-climbing exercise increased mitochondrial biogenesis within the cardiac muscle of middle-aged obese rats. Although there was no difference in the mitochondrial biogenesis-promoting mechanism (i.e., the PGC-1α protein level) among the different groups, 8 weeks of the ladder-climbing exercise significantly reduced ER stress-related factors, which are in turn biogenesis-inhibiting factors (Fig. 5). Although our data are not sufficient to draw any conclusion on these effects at this point, at least for the cardiac muscle, the results nevertheless indicate that concurrent stimulation from exercise training is more effective in protecting the cardiac muscle compared to a weight loss scheme that is entirely dependent on diet and reduced caloric intake. However, in this study, we used obese, middle-aged rats rather than rats with cardiac defects. Therefore, future studies focusing on subjects or models with cardiac defects should help to provide a clearer explanation of the effects of exercise training and energy control diet on protection of the cardiac muscle.



 XML Download
XML Download