

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cancer cells require huge amounts of nutrients to grow and survive. Amino acids are essential nutrients and transported into cells via selective transporters on the plasma membrane [12]. System L transporter transports neutral amino acids (valine, isoleucine, leucine, pheylalanine, tyrosine) into cells in a sodiumin-dependent manner [12].
Since Kanai et al. cloned the first L-type amino acid transporters (LAT1) [3], LAT2, LAT3, and LAT4 have been identified sequentially [456]. Interestingly, LAT1 and LAT2 have to form a heterodimer complex with an additional single-membrane-spanning protein, a heavy chain of 4F2 antigen (4F2hc), to perform their function as a transporter in the plasma membrane [34]. On the other hand, LAT3 and LAT4 can function as a transporter without 4F2hc [56].
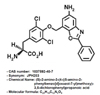
Since it has been known that LAT1 is highly and selectively expressed in a variety of cancer cells, LAT1 has become a novel target for cancer chemotherapy [378]. Compound 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH) was used to inhibit LAT1 but BCH inhibited both LAT1 and LAT2 [349]. The inhibition of LAT2 by BCH is problematic because LAT2 inhibition can disturb normal cell function and damage normal cell [1011]. Recently a selective LAT1 inhibitor named JPH203 ((S)-2-amino-3-(4-((5-amino-2-phenylbenzo[d]oxazol-7-yl)methoxy)-3,5-dichloropheyl) propanoic acid, Fig. 1) has been developed [12]. JPH203, a novel tyrosine analog, inhibits LAT1 selectively without affecting other types of LAT and has demonstrated strong inhibitory effects on the growth of various cancer cells in vitro [12131415].
Osteosarcoma is the most commonly diagnosed primary bone malignancy, in which the neoplastic mesenchymal cells show osteoid production [1617]. Recently it has been reported that LAT1 and CD98 (4F2hc) highly are highly expressed in human bone tumor like osteosarcoma and osteoblastoma [18].
This study examined the effects of JPH203, a novel tyrosine analog with high LAT1 selectivity, on cell growth, as well as its mechanism for cell death in Saos2 human osteosarcoma cells. The results demonstrated that JPH203 inhibits L-leucine uptake, cell growth, and colony formation in Saos2 human osteosarcoma cells, and further showed that JPH203 induces mitochondriadependent apoptotic cell death. Overall, this study provides evidence of the potential development of LAT1 inhibitor as a novel anticancer drug.
METHODS
Materials
[14C]L-leucine was purchased from PerkinElmer Life Sciences Inc. (Boston, MA, USA). BCH and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich Chemical Company (St Louis, MO, USA). Anti-caspase-3, anti-caspase-9, anti-poly (ADP-ribose) polymerase (anti-PARP), anti-p53, anti-Bcl-2, anti-Bcl-xL, anti-Bax, anti-Bak, anti-Bad, and anti-β-actin antibodies were supplied by Cell Signaling Technology, Inc. (Danvers, MA, USA). JPH203 HCl salt (purity>99%) was a kind gift from J-Pharma (Tokyo, Japan; Fig. 1). All other reagents were of analytical grade.
Cell lines and cell culture
Saos2 human osteosarcoma cells and FOB human osteoblastic cells were provided by American Type Culture Collection (ATCC; Rockville, MD, USA). Saos2 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS. FOB cells were grown in 1:1 mixture of Ham's F12 medium and DMEM without phenol red supplemented with 2.5 mM L-glutamine, 0.3 mg/ml G418, and 10% FBS. The cells were maintained as monolayers in plastic culture plates at 37℃ in a humidified atmosphere containing 5% CO2.
Quantitative real-time PCR (qRT-PCR)
For qRT-PCR analysis, total RNAs were prepared from Saos2 and FOB cells using TRIzol reagent (Gibco BRL, Grand Island, NY, USA), and quantified by spectrophotometry (Nanodrop 2000, Thermo Fisher Scientific Inc., Waltham, MA, USA) following the manufacturer's instructions. After isolation of total RNA, reverse transcription was carried out with 1 µg of total RNA using the ThermoScript RT-PCR system (Invitrogen, Carlsbad, CA, USA) for first strand cDNA synthesis according to the manufacturer's protocol. For qRT-PCR, cDNA were amplified by 2X TOP-simple™ DyeMIX-nTaq (Enzynomics, Seoul, Republic of Korea) using a SureCycler 8800 (Agilent Technologies, Santa Clara, CA, USA). Gene induction was determined using agarose gel electrophoresis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control for normalization. The deviations in the samples represent 4 separate experiments. For LAT1, the forward and reverse primers were 5′-CAAGGACATCTTCTCCGTCATC-3′ and 5′-AGCCACTTGGGCTTGTTT-3′, respectively. For LAT2, the forward and reverse primers were 5′-AAAGGGAGTGCTGGAGAATG-3′ and 5′-GACCCATGTGAGGAGCAATAA-3′, respectively. For 4F2hc, the forward and reverse primers were 5′-GGTGGAGCTGAATGAGTTAGAG-3′ and 5′-CGACATCATCCTTCTGGTTCTT-3′, respectively. For GAPDH, the forward and reverse primers were 5′-CTTTGGTATCGTGGAAGGACTC-3′ and 5′-AGTAGAGGCAGGGATGATGT-3′, respectively.
Transport measurements in cells
To examine inhibition of amino acid transport by JPH203 and BCH in Saos2 and FOB cells, uptake experiments were performed using [14C]L-leucine, which is a prototypical system L substrate and a model neutral amino acid, as described previously [3]. To determine the IC50 values of JPH203 and BCH, the uptake of 1.0 µM [14C]L-leucine was measured in the presence of JPH203 (0-100 µM) and BCH (0-3,000 µM), respectively.
MTT assay
The cell viability test was performed as described previously [910]. Saos2 and FOB cells were seeded at a concentration of 5×103 cells/well in 24-well plates. After 24 h of growth, the cells were treated with JPH203 or BCH at a range of concentrations and incubation times. The cell viability was assessed using a MTT assay. Three or four separate experiments were performed for each concentration/exposure time combination.
Colony formation assay
Colony formation assays were performed by seeding 300 cells/well into 6 well plates. After 24 h of growth, the cells were treated with 100 µM JPH203 for 72 h. The JPH203 treatment was removed and fresh medium was added. The cells were incubated for 10 days. Thereafter, medium was removed and the cells were washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde for 10 min at 4℃. Sequentially, the colonies were stained with 2% crystal violet for 10 min. Finally, colonies stained by crystal violet were washed with PBS and dried at room temperature, before imaged by a digital camera (PowerShot G12, Canon, Japan).
Annexin V-fluorescein isothiocyanate (V-FITC), propidium iodide (PI) staining, and flow cytometric analysis
Apoptosis was determined using an Annexin V-FITC assay. Saos2 cells were cultured in 100-mm tissue-culture dishes at a density of 1×106 cells/dish for 24 h and were treated with 0 or 3 mM JPH203 for 24 h. The cells were washed twice in phosphate-buffered saline and resuspended in a binding buffer (BD Biosciences, San Diego, CA, USA). Annexin V-FITC and 7-amino-actinomycin D (BD Biosciences) were added to the cells, which were then incubated in the dark for 15 min, and resuspended in 400 µl of binding buffer. The cells were analyzed using a fluorescence activated cell sorting Calibur flow cytometer (Becton-Dickinson, San Jose, CA, USA). Data analyses were performed using standard CellQuest software (Becton-Dickinson).
DNA fragmentation analysis
Following treatment with 0 or 3 mM JPH203 for 24 h, approximately 5×106 cells were collected and transferred into lysis buffer containing 100 mM NaCl, 10 mM EDTA, 300 mM Tris–HCl, pH 7.5, 200 mM sucrose, 0.5% SDS, and 0.5 mg/ml proteinase K and incubated at 65℃. DNA was extracted with an equal volume of phenol/chloroform/isoamylalcohol (25:24:1, v/v) and precipitated with ethanol. The DNA was resuspended in Tris–EDTA buffer, pH 8.0 containing 5 mg/ml DNase-free RNase and incubated at 37℃ for 1 h. The DNA was visualized on 2% agarose gel in the presence of 0.5 mg/ml ethidium bromide.
Immunoblotting
To determine the level of activated caspase-3, caspase-9, PARP, p53, Bcl-2, Bcl-xL, Bax, Bak, and Bad in the Saos2 cells treated with JPH203, the proteins were extracted, as described previously [11]. The anti-caspase-3, anti-caspase-9, anti-PARP, anti-p53, anti-Bcl-2, anti-Bcl-xL, anti-Bax, anti-Bak, anti-Bad and anti-β-actin antibodies were used as the primary antibody.
Data analysis
All experiments were performed at least in triplicate. The results were presented as the mean±S.E.M. Statistical significance was analyzed using a Student's t-test for two groups and one-way analysis of variance (ANOVA) for multi-group comparisons. A p-value<0.05 was considered a statistically significant difference.
RESULTS
Detection of system L amino acid transporters in Saos2 and FOB cells
In the real-time qRT-PCR analysis, PCR products for LAT1, LAT2 and their associating protein 4F2hc were detected in Saos2 and FOB cells (Fig. 2). In Saos2 (Fig. 2A) and FOB cells (Fig. 2B), the expressions for the products of LAT2 and LAT1, respectively, were very weak. The results from the qRT-PCR analysis indicate that LAT1 and LAT2 are present together with 4F2hc in Saos2 and FOB cells. And the expression of LAT2 in Saos2 cells is very weak, and the expression of LAT1 in FOB cells is very weak.
Inhibition of L-leucine uptake by JPH203 and BCH in Saos2 and FOB cells
As shown in Fig. 3A, JPH203 (0.001-100 µM) inhibited [14C]L-leucine (1.0 µM) uptake slightly in FOB cells with an IC50 value of 92.12±10.71 µM (mean±S.E.M., n=4 separate experiments). In contrast, JPH203 (0.001-100 µM) potently inhibited [14C]L-leucine uptake in a concentration-dependent manner in Saos2 cells with an IC50 value of 1.31±0.27 µM (n=4 separate experiments). As shown in Fig. 3B, BCH (1-3,000 µM) inhibited [14C]L-leucine uptake in Saos2 and FOB cells in a concentration-dependent manner with IC50 values of 72.17±0.92 µM (n=4 separate experiments) and 167.37±21.86 µM (n=4 separate experiments), respectively.
Inhibition of cell proliferation in Saos2 and FOB cells by JPH203 and BCH
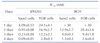
To examine the effect on cell viability, Saos2 and FOB cells were treated with JPH203 or BCH at various concentrations and incubation times, then analyzed using an MTT assay. As shown in Fig. 4A, when cells were treated with 0.01 to 30 mM JPH203 for 1-4 days, it potently inhibited cell proliferation in Saos2 cells in a dose- and time-dependent manner. In contrast, JPH203 only slightly inhibited proliferation of FOB cells (Fig. 4B). For BCH (dose range 0.1-30 mM), inhibition of cell proliferation in Saos2 cells was dependent on BCH treatment time and concentration (Fig. 4C). When FOB cells were treated with BCH, it inhibited cell proliferation in a dose- and time-dependent manner (Fig. 4D). Table 1 lists the IC50 values of JPH203 and BCH in Saos2 and FOB cells at set time points.
Suppression of colony formation in Saos2 cells by JPH203
A colony forming assay was used to determine whether JPH203 suppressed the proliferation ability of Saos2 cells. This assay revealed that colony formation was significantly inhibited by treatment with 100 µM JPH203 (Fig. 5A and 5B). Therefore, these results indicated that the proliferation of Saos2 cell was significantly suppressed by JPH203.
Apoptosis induction in Saos2 cells by JPH203
To determine if JPH203-induced cell death is associated with induction of apoptosis, Saos2 cells were treated with 3 mM JPH203 for 24 h and then co-stained with Annexin V-FITC, an apoptotic marker, and PI, a necrotic marker [13]. As shown in Fig. 6A, the percentage of Annexin V-FITC-positive cells in the early phase of apoptosis was increased 49.87% by JPH203 compared to the control. To determine if apoptosis is indeed the underlying mechanism for the observed reduced cell proliferation, Saos2 cells treated with JPH203 were subjected to DNA fragmentation assay. The formation of a DNA ladder was observed in Saos2 cells treated with 3 mM JPH203 for 24 h (Fig. 6B).
Mitochondria-dependent intrinsic apoptotic signaling pathway activated by JPH203 in Saos2 cells
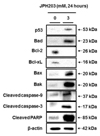
Next, to elucidate the apoptotic signaling pathways involved in JPH203-induced apoptosis in Saos2 cells, we measured the expression of apoptotic signaling pathway related proteins by western blotting in Saos2 cells treated with 3 mM for 24 h. The treatment of Saos2 cells with JPH203 increased the level of tumor suppressor p53 protein expression (Fig. 7). Furthermore, the expression levels of anti-apoptotic factors Bcl-2 and Bcl-xL were significantly decreased in Saos2 cells treated with JPH203, as shown in Fig. 7. Conversely, the expression levels of pro-apoptotic factors Bad, Bax, Bak, and cleaved caspase-9 were significantly increased by JPH203 (Fig. 7). Caspase-3 is a target molecule of mitochondria-dependent intrinsic apoptosis. Therefore, we analyzed the expression of cleaved caspase-3 and its downstream target molecule PARP. As shown in Fig. 7, cleaved caspase-3 was significantly upregulated in Saos2 cells treated with JPH203. Subsequently, the level of cleaved PARP, which functions downstream of caspase-3, was significantly increased by JPH203 treatment in Saos2 cells (Fig. 7). Taken together, these data indicate that JPH203-induced apoptosis in Saos2 cells was mediated by the activation of caspase-3 through the mitochondria-dependent intrinsic apoptotic signaling pathway.
DISCUSSION
In the previous study, we demonstrated the anti-cancer activity of JPH203, a newly developed selective LAT1 inhibitor, against YD-38 oral cancer cells which were originated from epithelial cells and were expressing only LAT1 [13]. In the present study, we tried expanding the therapeutic target for cancer cells by employing Saos2 human osteosarcoma cells which were originated from bone mesenchymal cells and were expressing both LAT1 and LAT2 [1617]. As the results, the findings indicated that JPH203 inhibited L-leucine uptake, cell growth, and colony formation in Saos2 human osteosarcoma cells, and further showed that JPH203 induced mitochondria-dependent apoptotic cell death.
Real-time qRT-PCR analysis (Fig. 2) showes that Saos2 and FOB cells express both LAT1 and LAT2, system L amino acid transporter isoforms, together with their associating protein 4F2hc. In Saos2 cells LAT2 expression was relatively poor, whereas in FOB cells LAT1 expression was relatively weak. Our results are consistent with previous report [19] indicating that Saos2 human osteosarcoma cells mainly expressed LAT1 with 4F2hc and showed weak LAT2 expression. To evaluate discriminative ability of JPH203 between Saos2 and FOB cells, we performed L-leucine uptake experiments in Saos2 and FOB cells.
In the present study, JPH203 potently inhibited L-leucine uptake in Saos2 cells (IC50 value; 1.31 µM), while slightly inhibiting it in FOB cells (IC50 value; 92.12 µM) (Fig. 3A). BCH inhibited L-leucine uptake in Saos2 cells (IC50 value; 72.17 µM) and FOB cells (IC50 value; 167.37 µM) (Fig. 3B). This means that JPH203 can distinguish relative abundance between LAT1 and LAT2. These results confirm that JPH203 has high selectivity for LAT1.
In the MTT assay, JPH203 potently inhibited cell growth in Saos2 cells but slightly inhibited growth in FOB cells (Fig. 4A, B and Table 1), and the affinity of JPH203 for this inhibition was higher in Saos2 cells than in FOB cells. BCH also inhibited cell growth in Saos2 and FOB cells (Fig. 4C, D and Table 1). In particular, the affinity of JPH203 for cell growth inhibition was higher than BCH in Saos2 cells, which corresponds to our results of the L-leucine uptake experiments. The apparent potency of JPH203 was >14-fold (2 days after compound treatment), >17-fold (3 days after compound treatment), and >14-fold (4 days after compound treatment) higher than BCH in Saos2 cells. Furthermore, in the colony forming assay, JPH203 suppressed the colony formation of Saos2 cells (Fig. 5), indicating that JPH203 significantly suppress the proliferation of Saos2 cell. These results suggest that cancer cell growth was preferentially inhibited by JPH203.
Although 100 µM JPH203 inhibited L-leucine uptake completely (IC50 value; 1.31 µM) in Saos2 cells, it was not enough to suppress cell growth (IC50 value; 90 µM) (Fig. 3A and Fig. 4A). This represents a 69-fold difference in susceptibility. Major concentration differences of JPH203 between uptake inhibition and growth arrest may be derived from the fact that the concentration of [14C]L-leucine was 1 µM in [14C]L-leucine uptake inhibition experiments, whereas the concentration of amino acids in culture media was much higher in growth inhibition experiments. The higher concentration of substrate for the transporter in growth inhibition study would need higher concentration of inhibitors to get a similar magnitude of inhibition. Yun et al. [13] and Oda et al. [12] reported similar results in YD-38 human oral cancer cells and HT-29 human colorectal adenocarcinoma cells, respectively. They also suggested that the differences in IC50 values are due to the amino acid concentrations (the substrate of LAT1) used in the incubation medium. In addition, their reports also suggested that JPH203 inhibits LAT1 by competing with its substrate [121320]. Therefore, the degree of inhibition is greatly influenced by the concentration of substrate.
It is difficult to clarify if growth arrest would translate to cytotoxicity, especially because cell viability was determined by MTT assay in this present study. To delineate the apoptotic process, we treated Saos2 cells with 3 mM JPH203 for 24 h (Figs. 6, 7). In the viability assay, treatment with 3 mM JPH203 for 24 h decreased the viability to about 50% in the Saos2 cells (Fig. 4A). As shown in Fig. 6, apoptotic population in FACS experiments was approximately 50% in the Saos2 cells treated with 3 mM JPH203. These findings demonstrate that the degree of cell viability measured by MTT assay show almost the same degree of apoptosis measured by annexin V-FITC assay in the Saos2 cells. Furthermore, as shown in Fig. 6B, 3 mM JPH203 induces genomic DNA laddering, a hall mark of apoptosis, in the Saos2 osteosarcoma cells. Taken together, these data indicate consistently that the lower concentration of JPH203 can induce the apoptosis in the Saos2 osteosarcoma cells.
Apoptosis is regulated precisely by the death receptor-mediated extrinsic and the mitochondria-dependent intrinsic pathway [2122]. Therefore, to verify the JPH203-induced apoptotic signaling pathway in Saos2 cells, we examined the alteration of proand anti-apoptotic factors associated with apoptosis. Members of Bcl-2 protein family localize to the mitochondrial membrane, and modulate apoptosis by permeabilizing the mitochondrial membrane, leading to release of cytochrome c from mitochondria [232425]. As shown in Fig. 7, treatment of Saos2 cells with JPH203 decreased levels of Bcl-2 and Bcl-xL, anti-apoptotic factors associated with the intrinsic mitochondrial-dependent apoptotic signaling pathway [2627]. Moreover, the JPH203 triggered the mitochondria-dependent intrinsic apoptotic signaling pathway, which involves upregulation or activation of pro-apoptotic factors, such as Bad, Bax, Bak, and cleaved caspase-9 [2223], as shown in Fig. 7. However, the last stage of this mitochondria-dependent pathway is mediated by activation of caspase-3 and PARP [142526]. Levels of cleaved caspase-3 and cleaved PARP were significantly increased in Saos2 cells treated with JPH203 (Fig. 7). Finally, as a typical phenomenon of apoptosis, activated PARP induced DNA fragmentation via single-strand DNA breakage in the nucleus of Saos2 cells.
The molecular biological pathways underlying inhibition of cancerous growth typically involve tumor suppressors, such as p53 [272829]. In the present study, p53 level was higher in Saos2 cells treated with JPH203 than in the control (Fig. 7), indicating that p53 may mediate the JPH203-induced apoptosis of Saos2 cells. However, the complete mechanisms of JPH203-induced apoptosis in Saos2 cells are not understood. Further studies will be needed to reveal these cellular and molecular mechanisms precisely.
In conclusion, inhibition of the amino acid transporter LAT1 by JPH203 led to apoptotic cell death mediated by the mitochondria-dependent intrinsic apoptotic signaling pathway in Saos2 human osteosarcoma cells. This effect is induced by the intracellular depletion of essential neutral amino acids, such as L-leucine. To our knowledge, this study using human osteosarcoma cells is the first report to demonstrate the in vitro anti-osteosarcoma effects of JPH203, a selective LAT1 inhibitor. Future work will involve additional JPH203 in vitro and in vivo studies as well as its potential development as a chemotherapeutic agent for the management of osteogenic sarcoma.





 XML Download
XML Download