

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Propofol (2,6-di-isopropylphenol) become a widely used intravenous anesthetic because of its rapid onset, short duration of action, and rapid elimination. It triggers vasorelaxation of a number of systemic vascular beds, including the aortae [1], the coronary arteries [2], the renal arteries [3]. However, effects of propofol on the pulmonary vascular system remain unclear. Machala et al. [4] studied the hemodynamic effects of propofol, found that propofol increased the pulmonary mean arterial pressure and afterload on the right ventricle. Funayama and his group [5] combined high thoraco-cervical epidural and general anesthesia in dogs found that propofol altered neither the pulmonary mean arterial pressure nor the pulmonary vascular resistance, but did decrease systemic vascular resistance. Another study [6] compared vasorelaxation induced by propofol in the intrapulmonary artery (IPA) and tehe extrapulmonary artery (EPA) and concluded that the response was significantly greater in the EPA than the IPA at higher drug concentrations. We earlier found that propofol exerted a relaxation effect in isolated rat pulmonary arterial rings [7]. In contrast, propofol potentiated phenylephrine-induced vasoconstriction in both live dogs and pulmonary arterial rings isolated from dogs [89]. Thus, the effect of propofol and its mechanism of action may vary by species and the location of the studied vessels.
The eicosanoid thromboxane A2 (TXA2), originating from the lungs, is released principally by activated platelets and vascular smooth muscle cells, and is one of the most potent vasoconstrictive agents known. It has been shown that the plasma concentrations of TXA2 and its stable metabolite, thromboxane B2 (TXB2), increase after hip arthroplasty [10], pulmonary resection, liver transplantation [11], and carotid endarterectomy [12]. TXA2 and its metabolite are evident immediately after skin incision [13], indicating that TXA2-mediated vasoconstriction may be a principal mechanism underlying the development of perioperative ischemic events. We used the thromboxane analog U46619 to precontract isolated human intrapulmonary arterial rings, allowing us to observe the effects of propofol on pulmonary vascular tone, and to explore the possible perioperative mechanism of action of propofol.
METHODS
Human pulmonary artery preparation
All research programs involving the use of human tissue are approved and supported by the Ethics Committee of Guangdong General Hospital (No.GDREC 2014023H [R1]). This investigation conformed to the principles outlined in the Declaration of Helsinki.
After obtaining informed consent, human pulmonary artery samples from patients who had undergone surgery for lung carcinoma were abtained. No patient had any clinical evidence of hypertension, pulmonary hypertension, diabetes, or use of nonsteroidal anti-inflammatory drugs (NSAIDs) prior to operation. Arteries were carefully removed from macroscopically normal regions of diseased lungs and immediately stored in oxygenated Kreb's solution (containing, in mM): NaCl 119, KCl 4.7, CaCl2 2.5, MgCl2 1, NaHCO3 25, KH2PO4 1.2, and D-glucose 11.1; the preparations were maintained at 4℃ and transferred to the laboratory within 30 min of resection. Arteries were isolated in a dissecting chamber filled with Kreb's solution. Fat and connective tissue were carefully removed under binocular microscopy, and 2~5 vascular rings (2~4 mm in internal diameter, 3~4 mm in length) were prepared from each artery.
Measurement of isometric tension
Arterial rings were mounted between two ‘L’-shaped stainless steel hooks under a resting tension of 2 mN placed in a thermostatically controlled (37.0±0.5℃) organ bath, 5 mL in capacity, containing Kreb's solution continuously aerated with carbogen (95% O2+5% CO2). The arterial rings were equilibrated for 90 min with replacement of the bath solution every 20 min. Sometimes, we cautiously inserted a small forceps into the ring lumen and rolled the ring backward and forward to denude the endothelium. After equilibration, a 60 mM K+-solution was added to the bath and contractile forces recorded. The vascular rings were washed four times with Kreb's solution, at intervals of 5 min, to restore basal tension. Changes in vascular tone of amplitudes <3 mN were considered to reflect poor contractibility, and such arterial rings were discarded. The endothelial integrity of pulmonary arterial rings was confirmed by acetylcholine (ACh, 10–5 mol/L)-induced relaxation of at least 50% at the serotonin plateau. Endothelium removal was confirmed by the absence of a vasorelaxant response to ACh. A high-sensitivity force-displacement transducer (Multi Myograph System; Danish Myo Technology, Aarhus, Denmark) was used to measure changes in tension; all data were recorded using version 5.4.1 of dedicated software (Powerlab; AD Instruments, Bella Vista, NSW, Australia).
Effect of propofol on pulmonary arterial rings
Propofol (10, 30, 100, 300 µM) was added to endothelium-denuded and intact rings cumulatively and a cumulative concentration-response curve (CRC) for propofol was obtained.
Effect of propofol on pulmonary vasoconstriction induced by U46619
Propofol (10, 30, 100, 300 µM) was added to endothelium-denuded and intact rings preconstrated by 100 nM U46619 cumulatively and a cumulative concentration-response curve for propofol was obtained.
The role of cyclooxygenase metabolites in propofol-induced vasoconstriction
Propofol (10, 30, 100, 300 µM) was added to endothelium-denuded and intact rings preconstrated by 100 nM U46619 cumulatively. After washing out the drugs from the organ bath, similar experiments were performed after 30 min, with the indubation of 100 µM indomethacin (a potent cyclooxygenase inhibitor).
Effect of propofol on pulmonary vasoconstriction induced by a 60 mM high-K+ solution
Propofol (10, 30, 100, 300 µM) was added to rings preconstrated by 60 mM high K+ solution cumulatively and a cumulative concentration-response curve for propofol was obtained.
The role of Ca2+ on the vasodilation effects of propofol
The rings were exposed to Ca2+ free, EGTA (500 µM) containing modified Kreb's solution after first equilibrated in normal Krebs solution for another 30 min. Then incubated in a Ca2+-free 60 mM K+ solution to the baseline before cumulative addition of CaCl2 (0.01 to 3 mM). Thereafter propofol (0, 10, 30, 100 µM) was respectively pretreated 30 min before another cumulative addition of CaCl2 (0.01 to 3 mM) and the cumulative concentrationresponse curve (CRC) for CaCl2 was obtained.
Measurement of [Ca2+]i by laser confocal fluorescence microscopy
The human pulmonary arteries were immersed in ice-cold physiological saline solution (PSS) containing (mM): 130 NaCl, 5 KCl, 2.7 MgCl2, 10 glucose, and 10 HEPES (pH 7.4, adjusted with NaOH). After surrounding connective tissues removed, the arteries were cut opened to expose the endothelial surface, and the endothelium was removed by gentle rubbing with a cotton swab. The tissues were digested at 37℃ for 20~40 min in Ca2+-free PSS containing type II collagenase (4 mg/ml), papain (4 mg/ml), bovine serum albumin (1 mg/ml), and DTT (0.5 mg/ml) [14]. The digestion was stopped by washing the tissue with PSS. The pulmonary arterial smooth muscle cells (PASMCs) were stored at 4℃ until experiments were performed.
PASMCs were incubated with 10 µM calcium indicator fluo-4AM in PSS at 37℃ for 30 min. The PSS was then replaced with Ca2+-free 60 mM K+-containing physiological saline solution containing (mM): 85 NaCl, 60 KCl, 2 EGTA, 1 MgCl2, 10 glucose, and 5 HEPES (pH 7.40, adjusted with NaOH). The cells were placed in an organ chamber at 37°C. The cells were treated with propofol (10~300 µM) and DMSO (control) for 10 min before fluorimetric measurements.
Fluorimetric measurements were performed using an Leica SP5-FCS laser scanning confocal system (Leica, Wetzlar, Germany) (excitation at 488 nm, emission filter at 525 nm). Calcium influx was trigged by addition of 2 mM CaCl2. Continuous recording of fluorescence images were obtained every 20 sec. Changes in [Ca2+]i was indicated by comparing the fluorescence intensity at a specific time point (F1) to that measured at the starting point of image recording (ΔF/F1*100, ΔF=F1–F0).
Chemicals
9,11-dideoxy-11a, 9a-epoxy-methanoprostaglandin F2a (U46619), indomethacin, and 2,6-di-isopropylphenol (propofol), were purchased from Sigma–Aldrich (St. Louis, MO, USA). U46619 and propofol were dissolved in DMSO and the other materials were dissolved in distilled water. Further dilutions were made from stock solutions. All concentrations are expressed as the final molar concentrations in the bath solution.
Statistical analysis
Data are expressed as means±SEMs, with the numbers indicating the numbers of vessels obtained from different patients. Contractile responses are expressed as percentages of the maximal contraction induced by high-K+ (60 mM). Relaxation was calculated as the percentage reduction of the active force at the stable plateau level. Bifunctional effects are expressed as the percentages of change in the active force mediated by U46619 or high-K+ (60 mM) at the stable plateau. Emax represented the maximal response percentage. EC50 represented the negative logarithm of the vasoconstrictor concentration required to produce half of the maximal contraction. pD2 represented the negative logarithm of the vasodilation concentration required to produce half of the maximal contraction. Emax, EC50 and pD2 were determined by non-linear regression curve fitting (Graphpad Prism software, version 5.0). Effect curves were analyzed by non-linear curve fitting using Sigmaplot version 10.0 (Systat Software, Chicago, IL, USA).
One-way ANOVA followed by the LSD test was used for statistical analysis when more than two groups were compared. Individual concentration response curves were compared by two-way analysis of variance followed by Bonferroni post-hoc testing. All statistical analyses were performed using SPSS version 13.0 software. A p-value <0.05 was considered statistically significant.
RESULTS
Effect of propofol on pulmonary artery resting tension
Propofol (10, 30, 100, and 300 µM) had no direct effect on the resting tension of endothelium-intact or -denuded pulmonary arteries (data not shown).
Effect of propofol on pulmonary vasoconstriction induced by U46619
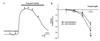
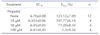
Propofol affected pulmonary rings contracted by U46619 in a biphasic manner. Propofol induced greater contractions in the endothelium-denuded group (EC50=4.699±0.12, Emax=31.19±5.10%) than in the endothelium-intact group (EC50=4.525±0.37, Emax=30.44±2.92%) at concentrations of 10-100 µM. At concentrations of 100-300 µM, propofol induced arterial contraction. The extent of contraction in the endothelium-intact and -denuded groups exposed to propofol concentrations of 10 and 30 µM differed significantly (both p levels <0.05; but groups exposed to 100 and 300 µM propofol did not differ significantly in this context (Fig. 1).
The role of cyclooxygenase metabolites in propofol-induced vasoconstriction
Indomethacin abolished contraction and simultaneously potentiated secondary relaxation. The maximal propofol-induced relaxation in the endothelium-intact group (pD2=3.713±0.11, Emax=98.72±0.34%) was higher than that in the endothelium-denuded group (pD2=3.54±0.03, Emax=94.56±0.53%). The endothelium-intact and -denuded groups did not differ significantly in terms of relaxation (p>0.05) when exposed to different concentrations of propofol (to 300 µM, commencing at 10-100 µM) (Fig. 2).
Effect of propofol on pulmonary vasoconstriction induced by a 60 mM-high-K+ solution
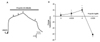
Propofol affected contraction of pulmonary rings in a 60 mM-high-K+ solution in a manner similar to U46619. Propofol induced arterial contraction (EC50=4.16±0.45, Emax=48.45±4.34%) from 10-100 µM, and relaxation from 100-300 µM (Fig. 3).
Propofol inhibited [Ca2+]i rise in human pulmonary smooth muscle cells
Addition of 2 mM CaCl2 triggered Ca2+ influx into human PASMCs in a Ca2+-free 60 mM-high-K+ solution. A 10 min incubation with propofol (10-300 µM) markedly reduced the maximal increase in [Ca2+]i, as indicated by fluorescent signaling from PASMCs, in a dose-dependent manner (Fig. 5).
DISCUSSION
We found that propofol exerted direct effects on isolated human pulmonary arteries and that the vascular effects noted depended on both vasomotor tone and propofol concentration. Propofol had no effect on baseline constriction in normal arteries but triggered marked pulmonary vasoconstriction when the vasomotor tone was acutely increased by addition of U46619. Propofol evoked constriction at low concentrations (10-100 µM), followed by secondary relaxation (100-300 µmol/L). Indomethacin abolished propofol-induced constriction. Propofol at various concentrations significantly inhibited the Ca2+-induced contraction of pulmonary rings exposed to Ca2+-free high-K+-containing solution, in a dose-dependent manner.
Only a limited number of studies have directly assessed the effects of propofol on the pulmonary circulation. This study is the first to use human pulmonary artery rings to explore the actions of propofol that precontracted by U46619. Rich et al. [14] found that, in contrast to the vasodilation produced by propofol in normal lungs, propofol significantly increased pulmonary vascular resistance after endothelial injury induced by electrolysis, suggesting that an endothelium-dependent mechanism was involved in the vasodilator response. In the cited work, the endothelium did not appear to mediate the contractile response; denudation of the endothelium potentiated the constrictions induced by propofol. In study on effects of propofol induction on haemodynamics, showed that propofol had no significant effect on either the mean pulmonary arterial pressure or pulmonary vascular resistance [4]. In elderly patients, propofol has been reported to trigger a transient increase in pulmonary vascular resistance, although the effect was not sustained during the entire infusion of propofol. Thus, propofol does not appear to exert a sustained effect on the baseline pulmonary circulation in humans. In contrast, propofol caused marked pulmonary vasoconstriction when the vasomotor tone was acutely increased by addition of U46619, which induces receptor-mediated activation of phospholipase C [15]. Stimulation of the associated signaling pathway increases the release of arachidonic acid, which is metabolized via the cyclooxygenase pathway to produce prostacyclin. Thromboxane A2, and other prostaglandins, with the aid of specific terminal synthases. Prostaglandins and thromboxanes activate smooth-muscle TP receptors (triggering vasoconstriction) and prostacyclin activates smooth-muscle IP receptors (triggering vasodilation) [16].
Indomethacin (a nonselective cyclooxygenase inhibitor) significantly reduced the propofol-induced contraction of both endothelium-intact and -denuded arteries, confirming that such constriction may be induced by cyclooxygenase-produced prostaglandins or by inhibiting the concomitant production of prostacyclin. The inhibition was endothelium-independent. Ogawa et al. [9] found that propofol potentiated alpha-adrenoreceptor-mediated pulmonary vasoconstriction by inhibiting the concomitant production of prostacyclin by cyclooxygenase. Cyclooxygenase has two isoforms, COX-1 and COX-2. COX-1 is expressed constitutively and produces physiological levels of prostaglandins and thromboxanes. In contrast, COX-2 is responsible for the increase in prostanoid production evident in pathological states. Long-term hypoxia enhanced COX-2 induction in unstimulated human PASMCs and significantly increased PGE2 release; COX-2 may play an important role in hypoxia-induced pulmonary hypertension [17]. Bradykinin also induces COX-2 expression in human PASMCs, in turn increasing prostaglandin production [18]. We found, in the present study, that incubation with indomethacin abolished the vasoconstriction of human pulmonary arteries induced by propofol. We speculate that propofol induced vasoconstriction by activating cyclooxygenase after U46619 had induced receptor-mediated activation of phospholipase C and stimulated the associated signaling pathway, thus increasing the release of prostanoids. This explains why propofol has no effect on baseline constriction in normal arteries but causes marked pulmonary vasoconstriction when the vasomotor tone is acutely increased by addition of U46619. Further, this response was endothelium-independent. However, the nature of the prostanoids requires further study.
The mechanisms of propofol-induced vasodilation vary by species and location. Tanaka et al. [6] found that vasodilatory responses to higher concentrations of propofol were greater in the rat extrapulmonary artery (EPA) than the intrapulmonary artery (IPA). A nitric oxide synthase inhibitor (L-NAME) decreased relaxation of the EPA, but had no effect on the IPA. Our team concluded that no significant difference of Emax was observed in the absence or presence of L-NAME in the rat IPA, the vasodilations were endothelium independent [7]. In the present study, the maximal vasodilator response was higher in endothelium-intact than -denuded rings, but relaxation of endothelium-denuded rings remained evident, showing that the relaxation response may partly depend on the endothelium. Ca2+ plays a very important role in vascular smooth muscle constriction. When the extracellular K+ concentration is elevated, the voltage-operated calcium channels of vascular smooth muscle cells are activated by membrane depolarization, increasing the [Ca2+]i and triggering vascular constriction. We found that propofol at different concentrations significantly inhibited the Ca2+-induced contractions of pulmonary rings exposed to Ca2+-free high-K+-containing solution, in a dose-dependent manner, indicating that propofol inhibited extracellular Ca2+ influx. In addition, propofol induced relaxation of arteries precontracted by 60 mM KCl solution, reduced of the rise in [Ca2+]i and the amplitude in human PASMCs as indicated by fluo-4 upon the application of CaCl2 to Ca2+-free high K+-containing solution suggests that the relaxant effects of propofol are mediated mostly by L-type voltage-operated calcium channels. in human PASMCs, as indicated by the extent of fluo-4 fluorescence upon addition of CaCl2 to Ca2+-free high-K+-containing solution. These data suggest that the relaxant effects of propofol are mediated principally by L-type voltage-operated calcium channels.
In this study our data show that propofol (100-300 µM) significantly inhibited the Ca2+-induced contraction of pulmonary rings exposed to high-K+-containing but Ca2+-free solution. Another experiment show that incubation with propofol (10-300 µM) reduced the maximal increase in [Ca2+]i, in a dose-dependent manner, especially at the concentration (100-300 µM) markedly and absolutely reduced the fluorescent signaling from PASMCs. So we speculate that propofol evoked constriction at low concentrations (10-100 µM) by promoting the concomitant production of prostaglandin by cyclooxygenase in PASMCs. When the concentration comes to a higher level (100-300 µM), the effect of propofol inhibiting calcium influx of PASMCs will take an advantage, result in a secondary relaxation.
In summary, unlike previous studies showing that propofol affected arteries in a single manner (relaxation or contraction), the present study demonstrates that propofol exerted a bifunctional effect on isolated human pulmonary arteries contracted by U46619. Propofol potentiated U46619-mediated pulmonary vasoconstriction by promoting the concomitant production of prostaglandin by cyclooxygenase in PASMCs, and relaxed arterial rings in a manner partly dependent on the endothelium, principally by inhibiting calcium influx through L-type voltage-operated calcium channels.




 XML Download
XML Download