

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammation is a defense phenomenon that protects injured tissues and organs from infectious pathogens such as viruses, fungi, and bacteria, as well as chemical toxins. In this process, innate immune cells such as macrophages, keratinocytes, and Langerhans cells are activated through Toll-like receptor (TLRs), which belong to a class of proteins that plays an essential role in activation of innate immunity [1]. Different TLRs respond to different pathogens [12]. For example, lipopolysaccharides (LPS), which are located in the outer membrane of Gram-negative bacteria, activate TLR4 expressed by macrophages and promote an inflammatory response by triggering intracellular signal transduction pathways [3]. Src, Syk, phosphoinositide 3-kinase (PI3K), AKT, and inhibitor of κB (IκB) kinase (IKK) α/β are major enzymes that are activated in macrophages upon LPS recognition by TLR4 [45]. Nuclear translocation of inflammation-regulating transcription factors such as nuclear factor (NF)-κB and activator protein (AP)-1 also occurs upon LPS stimulation of macrophages. These transcription factors upregulate the expression of genes encoding cytokines such as interleukin (IL)-1 and tumor necrosis factor (TNF)-α, chemokines, and inflammatory mediators such as inducible nitric oxide (NO) synthase (iNOS) (involved in NO production) and cyclooxygenase (involved in prostaglandin E2 secretion) [567]. A chronic and sustained inflammatory response is characteristic of serious inflammatory diseases including arthritis, cancer, asthma, diabetes, and cardiovascular disease [8910]. There is therefore great interest in discovering treatments that are not systemically immunosuppressive but selectively attenuate unnecessary inflammatory responses.
Beauvericin is a toxic cyclic hexadepsipeptide made up of alternating N-methyl-L-phenylalanine and hydroxy isovaleric acid residues that has been isolated from various fungi, including Beaveria bassiana and Fusarium spp. [1112]. BEA is a mycotoxin with a similar structure to enniatin that was first studied for its insecticidal properties [131415]. In addition, BEA has antimicrobial, anti-convulsion, anti-inflammatory, immunosuppressive, and anti-tumor activities [1617]. With regard to its anti-bacterial activity, BEA targets the synthesis of organelles and enzyme systems unlike other antibiotics, which block cell wall synthesis [1516]. BEA can suppress T cell activation by targeting PI3K/AKT signaling and can induce apoptosis in H4IIE hepatoma cells by inhibiting NF-κB activity and mitogen-activated protein kinases [1819]. However, few studies have examined the anti-inflammatory effects of BEA or explored the direct pharmacological targets of BEA. In this study, we therefore investigated the anti-inflammatory activity of BEA and the underlying molecular mechanisms using LPS-treated macrophages.
METHODS
Materials
Beauvericin (purity: 97%), 5-diphenyltetrazolium bromide (MTT), and lipopolysaccharide (LPS; E. coli 0111:B4) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). A luciferase construct that contained promoters sensitive to NF-κB was used as reported previously [20]. Fetal bovine serum (FBS), antibiotics (penicillin and streptomycin), phosphate-buffered saline (PBS), DMEM, RPMI 1640, and trypsin were obtained from Gibco (Grand Island, NY, USA). The murine macrophage cell line RAW264.7 and human embryonic kidney cells were purchased from the American Type Culture Collection (Rockville, CA, USA). PP2 and piceatannol were obtained from Calbiochem (La Jolla, CA, USA). Hoechst staining solution and antibodies (phospho-specific and/or total protein) against p65, p50, lamin A/C, p85, AKT, inhibitor of κBα (IκBα), Src, Syk, Myc, and β-actin were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody against HA was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rhodamine phalloidin was purchased from Life Technologies (Waltham, MA, USA).
Cell culture and drug preparation
RAW264.7 and HEK293 cells were incubated in DMEM or RPMI1640 medium supplemented with 10% fetal bovine serum and antibiotics (streptomycin: 100 µg/ml, penicillin: 100 U/ml), respectively, and grown at 37℃ and 5% CO2. Stock solution of BEA was prepared in dimethylsulfoxide (DMSO) and stored at
−70℃ until use.
Greiss assay for NO production
RAW264.7 cells were seeded (1×106 cells/ml) on a plate and preincubated for 18 h. Cells were pretreated with BEA (0 to 4.5 µM) or standard compounds (L-NAME, PP2, and piceatannol) for 30 min, and then LPS (1 µg/ml) was added for 24 h. The effects of BEA on NO production were determined by analyzing the NO level using Griess reagent, as described previously [2122].
Immunofluorescence and image analysis
RAW264.7 cells were plated on a glass coverslip. After 1 h, the cells were incubated with BEA (2 and 4 µM) for 30 min, and further treated with LPS (1 µg/ml) for 12 h. Cells were then fixed in 3.7% paraformaldehyde in PBS and then permeabilized with 1% Triton X-100 in PBS at room temperature. After washing, cells were treated 1% bovine serum albumin in PBS to minimize nonspecific staining. For staining, cells were incubated with rhodamine phalloidin conjugate (1:40 dilution) and Hoechst staining solution (1:1000 dilution) for 1 h at room temperature. DAPI was used for DNA counterstaining, and rhodamine phalloidin was used for specific filamentous actin counterstaining [2324]. A confocal laser-scanning microscope (LSM 700; Zeiss, German) equipped with a C-Apochromat 63× /1.2 water immersion objective was used to acquire images. A Nikon Eclipse Ti inverted microscope equipped with a CCD-cooled camera (DS-Qi1Mc, Nikon, Japan) and ZEN 2010 LSM software (Zeiss, Germany) was used for image analysis [25].
Cell viability test
The cytotoxicity of BEA was evaluated using the conventional MTT assay, as reported previously [2627]. To measure cytotoxicity, RAW264.7 cells were pre-incubated for 18 h (1×106 cells/ml), and then BEA (0 to 4.5 µM) was added to the cells, which were incubated for an additional 24 h. After 3 h of culture termination, 10 µl of MTT solution (10 mg/mL in phosphate buffered-saline, pH 7.4) was added to each well. To stop the MTT reaction and solubilize the formazan crystals, 15% sodium dodecyl sulfate (SDS) was added to each well. The absorbance at 570 nm (OD570–630) was measured on a Spectra Max 250 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
mRNA expression measurement by semi-quantitative RT-PCR
For mRNA analysis, RAW264.7 cells were pre-treated with BEA for 30 min and then further treated with LPS for up to 6 h. Expression of inflammation-related markers was evaluated by semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). TRIzol reagent (Gibco BRL) was used to isolate RNA from LPS-treated RAW264.7 cells. Total RNA was stored at −70℃ until use. Semi-quantitative RT-PCR reactions were conducted as reported previously [728]. The primers (Bioneer, Daejeon, Korea) used in these reactions are listed in Table 1.
DNA transfection and luciferase reporter gene activity assay
For transfection of plasmids (MyD88 or TRIF) into HEK293 cells and LPS-stimulated RAW264.7 cells, the cells (1×106 cells/ml) were seeded in a 12-well plate and transfected with 1 µg of plasmids using PEI method [29]. To overexpress Src and Syk, HA-Src and Myc-Syk plasmids were transformed into HEK293 cells. For the luciferase assay, HEK293 cells and RAW264.7 cells were transformed with NF-κB-Luc along with β-galactosidase and inducing molecules such as TRIF or MyD88. Luciferase assays were performed using Promega's Luciferase Assay System (Promega, Fitchburg, WI, USA), as reported previously [30].
Preparation of cell lysates for nuclear fractions and immunoblotting
RAW264.7 and HEK293 cells (5×106 cells/ml) were washed in cold PBS containing 1 mM sodium orthovanadate and then lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 2 mM EDTA, 2 mM ethylene glycol tetraacetic acid, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM dithiothreitol, 1% Triton X-100, 10% glycerol, 10 µg/ml aprotinin, 10 µg/ml pepstatin, 1 mM benzimide, and 2 mM PMSF) for 30 min with rotation at 4℃. Cell lysates were centrifuged at 16,000×g for 10 min at 4℃ and stored at −20℃ [31]. Nuclear lysates of RAW264.7 cells were prepared using a three-step procedure, as reported previously [32]. In the first step, cells were treated with BEA and LPS, collected with a rubber policeman, washed with PBS, and lysed in lysis buffer (50 mM KCl, 0.5% Nonidet P-40, 25 mM HEPES (pH 7.8), 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 20 µg/ml aprotinin, and 100 µM 1,4-dithiothreitol (DTT)) on ice for 4 min. Next, cell lysates were centrifuged at 19,326×g for 1 min in a microcentrifuge. In the second step, the pellet (nuclear fraction) was treated with wash buffer (lysis buffer without Nonidet P-40) and resuspended three times. Last, nuclei were resuspended in extraction buffer (lysis buffer to which 500 mM KCl and 10% glycerol were added). Nuclei/extraction buffer mixtures were frozen at −80℃ and centrifuged at 19,326×g for 5 min. Supernatants were collected as the nuclear extract. Soluble cell lysates (nuclear and whole cell lysates) were analyzed by immunoblotting, and protein levels were visualized as reported previously [33].
Statistical analysis
Data are expressed as mean±standard deviation (SD) of at least three different experiments with similar results. For statistical comparisons, variance/Scheffe's post-hoc tests and Kruskal-Wallis/Mann Whitney tests were used. All p values <0.05 were considered statistically significant. All statistical tests were implemented using the computer program SPSS (SPSS Inc. Chicago, IL, USA).
RESULTS
Effect of BEA on inflammatory responses of macrophages
To determine the anti-inflammatory effect of BEA on LPS-exposed RAW264.7 cells, cells were pretreated with different doses of BEA (0, 3.5, 4, and 4.5 µM). NO levels in the culture supernatants of BEA-treated cells were then determined. As Fig. 1A shows, BEA dose-dependently diminished secreted levels of NO (almost 50% decrease at 4.5 µM). The morphology of activated RAW264.7 cells was also strongly affected by BEA treatment at 2 and 4 µM (Fig. 1B). To investigate whether the suppressive activity of BEA on NO production and morphology was due to cytotoxicity, cell viability was examined using a conventional MTT assay. As Figs. 1C and 1D display, BEA (up to 7.5 µM) did not significantly affect the viability of RAW264.7 or HEK293 cells after 9 or 12 h.
Effect of BEA on the transcriptional regulation of inflammatory genes in LPS-treated RAW264.7 cells
To investigate how BEA decreased NO production, we determined mRNA levels of inflammatory genes in LPS-stimulated RAW264.7 cells. After treatment of RAW264.7 cells with BEA (5 and 7.5 µM) for 30 min, cells were further stimulated with LPS (1 µg/ml) for 6 h. Transcript levels of IL-1β and iNOS were dramatically suppressed by BEA (7.5 µM) (Fig. 2A). In a luciferase reporter gene assay, BEA diminished NF-κB-mediated luciferase activity induced by cotransfection of HEK293 cells with a plasmid encoding myeloid differentiation primary response 88 (MyD88) but not TIR-domain-containing adapter-inducing interferon-β (TRIF) (51% decrease at 7.5 µM of BEA) (Fig. 2B). This compound (7.5 µM) also diminished NF-κB-mediated luciferase activity induced by LPS in MyD88-transfected RAW264.7 cells up to 40.2% (Fig. 2C). Finally, the effect of BEA on the nuclear translocation of NF-κB subunits (p50 and p65) was elucidated by immunoblot analysis of nuclear fractions of LPS-stimulated RAW264.7 cells exposed to BEA. As Fig. 2D shows, nuclear levels of p50 and p65 were reduced by BEA (7.5 µM) at 30 and 60 min in LPS-stimulated RAW264.7 cells.
Effect of BEA on upstream signaling events regulating NF-κB translocation in LPS-stimulated RAW264.7 cells
Because BEA clearly suppressed translocation of NF-κB subunits in LPS-activated RAW264.7 cells, we next evaluated the regulatory role of BEA on the activation of signaling events upstream of NF-κB translocation. After preparing whole cell lysates of BEA-treated RAW264.7 cells stimulated with LPS, phosphorylated forms of upstream signaling proteins were analyzed by immunoblot analysis. As shown in Fig. 3A, levels of phospho-IκBα, phospho-p85/PI3K, and phospho-ATK were reduced by BEA (7.5 µM) at 15, 30, and 60 min (Fig. 3A). Because the LPS-induced increase in levels of phospho-p85 at 5 min was decreased by BEA (Fig. 3A), we examined whether BEA was able to suppress the activity of Src and Syk, enzymes that phosphorylate p85/PI3K, by measuring the phospho-forms of Syk and Src by immunoblot analysis. As Fig. 3B shows, BEA (7.5 µM) strongly inhibited phosphorylation of Src and Syk at 2, 5, 10, and 15 min (Fig. 3B), implying that these enzymes are targets of BEA.
Effect of BEA on the phosphorylation of PI3K/p85 induced by Src or Syk in HEK293 cells
To investigate whether Src and Syk are pharmacological targets of BEA, we overexpressed Src or Syk constructs and measured levels of phospho-p85 in HEK293 cells using immunoblot analysis. As shown in Figs. 4A and 4B, overexpression of Src and Syk increased the level of phosphorylated p85. As expected, BEA (7.5 µM) clearly reduced the Src- and Syk-induced increases in phosphorylated p85 (Figs. 4A and 4B). Using Src and Syk inhibitors (PP2 and piceatannol, respectively), we confirmed that Syk and Src are important regulators of the inflammatory responses by determining phospho-p85 levels and NO secretion in LPS-treated RAW264.7 cells. PP2 at 10 and 20 µM strongly inhibited the LPS-induced increase in phospho-p85 levels (Fig. 4C). In addition, PP2 and piceatannol strongly blocked the release of NO from LPS-treated RAW264.7 cells (Fig. 4D).
DISCUSSION
BEA has previously been reported to exhibit anti-inflammatory activity in an experimental colitis model by inhibiting activated T cells [18]. However, little is known about how the anti-inflammatory actions of BEA are mediated [16]. Because macrophages are major inflammatory cells activated by LPS, a TLR4 ligand [23], we tested the inflammatory responses RAW264.7 cells to BEA treatment to elucidate the anti-inflammatory activity of BEA and its underlying molecular mechanisms. As shown in Figs. 1 and 2, BEA was a strong anti-inflammatory agent that inhibited NO production, inflammatory gene expression, and LPS-induced morphological changes in macrophages. By evaluating the transcriptional activation of NF-κB, we found that the anti-inflammatory activity of BEA was mediated by a transcriptional level effect on inflammatory genes, as seen in the case of apoptosis-inducing activity in H4IIE hepatoma cells [19]. Interestingly, numerous compounds and plant-derived extracts, including 4-isopropyl-2,6-bis(1-phenylethyl)aniline 1, ginsenosides, scutellarein, and Phyllanthus acidus, all of which have strong anti-inflammatory activity, have been identified as NF-κB pathway inhibitors [3435363738]. Our data and these reports strongly suggest that NF-κB is the primary mediator of macrophage-inflammatory responses, as reported previously [394041]; suppression of this factor could be responsible for the strong anti-inflammatory effects of BEA and the other compounds described above.
To understand how BEA suppresses the nuclear translocation and activation of NF-κB (Fig. 2D), we examined upstream signaling events involved in regulation of NF-κB translocation. As Figs. 3 and 4 show, phosphorylation of IκBα, AKT, phosphatidylinositol 3-kinase (P13K), Src, and Syk play a critical role in the activation of NF-κB [4243]. We found that BEA suppressed the activation of these enzymes (Figs. 3 and 4), implying that these enzymes could be targeted in BEA-mediated suppression of the NF-κB pathway. In particular, the phosphorylation of p85 induced by Src and Syk overexpression was strongly suppressed by BEA, indicating that this compound can directly suppress Src and Syk. Moreover, the finding that MyD88-dependent activation of NF-κB in both HEK293 and LPS-stimulated RAW264.7 cells was inhibited by BEA (Fig. 2B and 2C) suggested that activation of Src and Syk might be preferentially induced by a MyD88-mediated signaling cascade. Several reports support this possibility in that MyD88, but not TRIF is associated with Src or Syk via IRAK1/4 [4445]. Based on these findings, BEA may be more effective against TLR2- and TLR4-mediated inflammatory responses than TLR3-mediated responses, because TLR3 is not associated with MyD88 [46]. Thus, BEA may have a greater antiinflammatory effect in bacterial infection-derived inflammatory diseases than viral infectious diseases as shown in the cases of ginseng saponins. This hypothesis requires further testing in relevant in vivo animal models, including mouse models of septic shock and influenza virus infection.
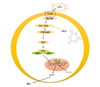
In summary, we demonstrated that BEA is able to dose-dependently suppress inflammatory responses such as NO production, morphological changes, and inflammatory gene expression in LPS-treated RAW264.7 cells. BEA is capable of inhibiting the nuclear translocation of the NF-κB subunits p65 and p50 and suppresses the activation of the upstream enzymes Src and Syk as summarized in Fig. 5. These results strongly suggest that BEA is a strong anti-inflammatory agent that attenuates NF-κB-dependent inflammatory responses by inhibiting both Src and Syk. In future pre-clinical studies, we intend to examine the anti-inflammatory activity of BEA in mice and to elucidate the underlying molecular mechanisms.





 XML Download
XML Download