

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The preterm birth is a process frequently initiated by a uterine infection and is accompanied by an increase in pro-inflammatory cytokines such as TNF-α or IL-1β [12]. The increase of these pro-inflammatory cytokines favors the stimulation of uterine contractions and induces the onset of labor [34]. This inflammatory response provokes the release of uterotonic agents, such as PGF2α that stimulates the uterine contraction and is a well-recognized smooth muscle motility inducer [5]. On the contrary, many of the uterus relaxant mechanisms involve the second messenger cyclic adenosine monophosphate (cAMP); in fact, cAMP is responsible for the signal transduction that stimulates the cAMP-dependent pathway, such as the activation cAMP-dependent protein kinase (PKA) that, in turn, phosphorylates and inhibits key proteins which cause uterine contractions [6]. The search for new ways to increase the cAMP concentration in smooth muscle is an area of interest; thus, the inhibition of phosphodiesterase-4 (PDE-4) has been suggested as an pharmacological target [7]. PDE-4 is responsible for cAMP-hydrolysis and is predominantly expressed in the smooth muscle of myometrium and airways as well as in immune-associated cells, such as monocytes and neutrophils [8910]. Therefore, the development of effective inhibitors of PDE-4 may lead to new myorelaxant and anti-inflammatory drugs [1112]. PDE-4 has four subtypes (PDE-4A to PDE-4D), the PDE-4B subtype is overexpressed in near-term pregnant myometrium as well as in infection-induced premature delivery [13]. Furthermore, it has been shown that the regulation of the PDE-4B2 isoform in human uterine cells is related to the local release of pro-inflammatory cytokines and uterotonic agents, while selective PDE-4-inhibitors negatively regulate the activation of the immune cells and pro-inflammatory cytokines release [14].
The β-mimetics salbutamol and terbutaline have been used to inhibit premature uterine contractions as they increase the concentration of cAMP through the activation of adenylyl cyclase (AC) which synthesizes cAMP [15]; however, their efficacy has been questioned because of serious fetal or maternal side-effects [16]. Therefore, the inhibition of PDE-4 to increase the cAMP has been suggested as a more specific and safer pharmacological approach for the preterm birth treatment. PDE-4 inhibitors such as rolipram, cilomilast, and roflumilast have been studied in diverse preterm birth models [71217]. In fact, a family of non-teratogenic thalidomide analogs are potent PDE-4 inhibitors as well as they are more effective as immunomodulatory and anti-inflammatory agents because they inhibit TNF-α significantly, such as apremilast [61819202122]. A prior study in pregnant human myometrium demonstrated that two thalidomide analogs, methyl 3-(4-nitrophthalimide)-3-(3,4-dimethoxyphenyl)-propanoate (4NO2PDPMe) and methyl 3-(4-aminophthalimido)-3-(3,4-dimethoxyphenyl)-propanoate (4APDPMe), were promising uterus-relaxant agents because of their potency and efficacy [6]. Hence, to corroborate such properties on pharmacological models emulating infection-induced contractions, as those caused by the uterotonic PGF2α, might result interesting. Besides, to assess their immunomodulatory and anti-inflammatory effects as PDE-4 inhibitors on pro- and anti-inflammatory cytokines induced by lipopolysaccharides (LPS) in uterus has not been previously reported. Furthermore, in that preceding study it was suggested that these thalidomide analogs and rolipram may also be calcium-channel blockers; thus, to verify it and to compare their effects to nifedipine which is used for the treatment of preterm labor, seemed desirable. Therefore, the aim of the present study was to evaluate the relaxant and anti-inflammatory effects of two thalidomide analogs, 4NO2PDPMe and 4APDPMe, as PDE-4 inhibitors in pregnant rat uterus.
METHODS
Animals
Ninety pregnant female Wistar rats weighing 200~220 g (70~80 days old) were used. Pregnancy was corroborated daily and the female rat was catalogued as pregnant when a copulation plug was observed in the vagina; the presence of the plug was considered as the first day of pregnancy. Four animals per cage were housed under the following conditions: temperature (20~25℃), humidity (40~60%), 12 h of light/12 h of dark and ad libitum water and food intake. All the animals received humane care according to the respective institutional guidelines, the Mexican Official Norm (NOM-062-ZOO-999) [23] regarding technical specifications for the production, care and use of laboratory animals, and the criteria outlined in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 1985).
Drugs and solutions
Thalidomide analogs, 4NO2PDPMe and 4APDPMe, were synthesized as previously described [624]. Rolipram (selective PDE-4 inhibitor), forskolin (direct activator of AC), nifedipine (calcium channel blocker), Escherichia coli LPS (serotype 055:B5), prostaglandin PGF2α, Dulbecco's modified Eagle's medium and Dulbecco phosphate buffered saline (PBS 10x), Dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Functional studies of in vitro contractility
Pregnant rats were sacrificed by CO2 inhalation at day 19 of gestation to study the relaxant effect of both thalidomide analogs as tocolytic agents. Uterus removal and its preparation for the in vitro experiments was carried out as reported [25]. Isolated uterine horns were placed in a Ringer physiological solution (containing in mM: NaCl 144, NaHCO3 30, KCl 6.2, KH2PO4 1, MgSO4 0.5, CaCl2 2.5, glucose 11.1, pH 7.4) bath to remove fetus-placental fragments. Uterine strips, 10×3 mm, were removed from the antimesium side of the pregnant uterus and vertically mounted in chambers with 3 mL of Ringer solution. After mounting, the Ringer solution was changed repeatedly until the basal tension record was equilibrated to 1 g. Tissues were maintained in the Ringer solution bath at 37℃ with constant bubbling of 5% CO2 in O2 before the contractility assays, which were initiated and induced by pharmaco- and electro-mechanic stimuli using PGF2α (1 µM), K+ (40 mM) and Ca2+ (1 mM). The changes of contractile activity from the isometric tension was recorded with a FT03C force transducer coupled to a RPS-312 RM model polygraph (Grass Telefactor, RI, USA). The data were analyzed using the software PolyVIEW version 2.5 and the uterine contractions (integral activity) were measured considering the area under the curve (AUC) defined by the graphic isometric record over a 20-min period after stabilization. The inhibitory effects of thalidomide analogs, rolipram, forskolin and nifedipine on phasic and tonic uterine contractions were expressed as follows:
AUCr is the remaining AUC after uterine tissue exposition to drug, and AUCi is the AUC of the integral activity prior to any compound addition. A period of 20 min, before and after exposition to drugs, was considered enough to obtain stable and representative biological activity. All concentration-response curves were generated for both analogs and rolipram using the following concentrations: 10, 32, 56, 86, 100, 180, 230 and 320 µM; for forskolin: 0.18, 0.56, 1, 1.8, 3.2 and 5.6 µM; and for nifedipine: 1, 3.2, 10, 18, 23 and 56 nM.
cAMP accumulation in pregnant rat uterus
Uterine tissues (100 mg) were washed four times in PBS (1×) to remove excess blood and then incubated for 2 h in 24-well plates, which contained 1 mL of Dulbecco's serum-free modified Eagle's medium supplemented with 100 U/ml penicillin and 100 µg/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA) at 37℃ in a humidified atmosphere with 5% CO2. After 2 h of incubation, the culture medium was changed and the uterine tissues were incubated first with the PDE-4 inhibitors at increasing concentrations of 100, 300 and 1,000 µM for 1 h and then with 6 µM forskolin for 30 min; these concentrations were selected from a former study [6]. All compounds were dissolved in 10 ml of DMSO. After the stimulation, the samples were collected, frozen in liquid nitrogen and stored at −80℃ until use as reported [123]. Frozen tissue samples were homogenized (100 mg/ml), with a tissue-tearor homogenizer (BioSpec Products Inc., Bartlesville, OK, USA) in ice-cold homogenization buffer (2 mM MgSO4, 2 mM EDTA, 1 mM β-mercaptoethanol, 100 mM Tris/HCl (pH 7.4) and 10% glycerol), supplemented with protease inhibitor cocktail as previously reported [13]. The cAMP-assay was performed following the instructions of the direct cAMP ELISA Assay kit (Enzo Life Sciences, Inc., Exeter, UK). cAMP concentrations were determined at 405 nm using a Multiskan EX plate reader (Thermo Scientific, Vantaa, Finland) and expressed as pg/ml.
Determination of anti-inflammatory effects
For cytokine measurement by enzyme-linked immunoassay experiments, the pregnant rat uterine explants were cut in small pieces of 50 mg and washed 4 times with 1× PBS and then placed in 24-well plates, which contained 1 mL of culture medium per well (Dulbecco's modified Eagle's medium) supplemented with 100 U/ml penicillin and 100 µg/ml streptomycin. The explants were incubated at 37℃ in a 5% CO2 humidified atmosphere for 3 h, the tissue samples were then transferred to new 24-well plates containing fresh culture medium. The uterine explants were incubated with increasing concentrations of 100, 300 and 1,000 µM of rolipram, 4ADPDMe, 4NO2PDPMe and 10 µM forskolin, with or without 10 µg/ml of LPS for 24 h, as recommended [226]. All compounds were dissolved in DMSO to rich a maximum final concentration of 10 µg/ml. Each experiment was carried out in duplicates. After the incubation period, the supernatants were collected and stored at −20℃, until use as reported [226]. The concentrations of TNF-α, IL-1β and IL-10 in the supernatants of undiluted culture medium were determined using rat TNF-α, IL-1β and IL-10 ELISA kits (Novex Life Technologies, Waltham, MA, USA); manufacturer's directions were followed and samples read using a Multiskan EX plate reader. Cytokine concentrations were expressed as pg/ml.
Statistical analysis
Data for the effect of drugs on phasic and tonic contractions underwent a concentration-response curve analysis, which was performed using Sigma Plot version 10 software (Systat Software Inc., San Jose, CA, USA) to obtain the inhibitory concentration-50 (IC50) values, a drug concentration that produces 50% of the maximum inhibitory effect (potency), and the Emax, a maximum inhibitory response that can be produced by the highest concentration of the tested compound (efficacy). Data are expressed as the means±SEM from determinations for each concentration (n=6). The differences in IC50 and Emax among the compounds were determined by one-way ANOVA followed by a post hoc Student-Newman-Keuls test using Sigma Stat software version 3.1 (Systat Software Inc., San Jose, CA, USA). In all cases, p<0.05 was considered statistically significant.
RESULTS
Thalidomide analogs inhibit phasic and tonic contractions of pregnant rat uterus
Fig. 1a shows the concentration-dependent sigmoid curves of the inhibitory effect on the PGF2α-induced phasic contractions of pregnant rat uterus by the PDE-4 inhibitors, thalidomide analogs and rolipram, which were compared to the Ca2+-channel blocker nifedipine and the direct AC activator forskolin. Nifedipine sigmoid curve totally appears leftward in the order of 10-8 M concentration and reached a maximal inhibition of 97.6%, while forskolin sigmoid curve is observed in the order of µM and its maximal inhibition was almost 80%, thus forskolin was less potent and effective than nifedipine; in addition, the non-parallel slope and shape of both sigmoid curves suggest the different mechanisms of action of these drugs. Sigmoid curves of thalidomide analogs and rolipram were found very close to each other and even more rightward in the order of 10-4 M, this indicates that PDE-4 inhibitors are less potent than the comparison drugs; moreover, their order of potency was as follows: 4APDPMe was the most potent>4NO2PDPMe>rolipram. Regarding the efficacy of PDE-4 inhibitors, 4APDPMe was also more effective than 4NO2PDPMe>rolipram (p<0.05), and as effective as nifedipine, because the maximal inhibition of 4APDPMe was 99.4%. Fig. 1b depicts a typical tracing of the inhibition of PGF2α-induced phasic contractions of pregnant rat uterus by thalidomide analog addition.
In fact, a very similar pattern in the distribution and shape of concentration-dependent sigmoid curves of the inhibitory effect on the K+- and Ca2+-induced tonic contractions of pregnant rat uterus was observed as can be seen in Fig. 2a and 3a, along with their representative typical recordings of the experimental inhibition of those tonic contractions by thalidomide analog addition (Fig. 2b and Fig. 3b, respectively). A summary of the pharmacological parameters IC50 and Emax values for both thalidomide analogs and rolipram, as well as for nifedipine and forskolin, are presented in Table 1, all of which were derived from the concentration-response curve analysis. Both uterine tonic contractions appeared to be more sensitive to the inhibitory effects of the PDE-4 inhibitors when compared with phasic contractions because their IC50 values were lower than the IC50 required during PGF2α-induced phasic contractions; thus, 4APDPMe was the most potent inhibitor of contractions among the PDE-4 inhibitors and was equieffective to nifedipine (practically 100% of inhibitory effect). Rolipram was the less potent because presented the highest IC50 values for the two types of contraction, while forskolin was the less effective since it achieved the lowest Emax values (p<0.05).
Thalidomide analogs increase cAMP levels in pregnant rat uterus
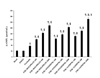
The direct activator of AC forskolin (6 µM) induced an almost two-fold increase in the uterine level of cAMP when compared to the basal amount (p<0.05), which was not modified by the vehicle DMSO used to dissolve the compounds (Fig. 4). However, the inhibition of PDE-4 due to the thalidomide analogs or rolipram augmented the uterine levels of the forskolin-induced cAMP in a concentration-dependent manner. Rolipram and 4NO2PDPMe elicited similar cAMP levels in the pregnant rat uterus with the three concentrations employed (100, 300 and 1,000 µM), while 4APDPMe increased that second messenger in a higher degree and even in a significant way compared with forskolin plus 1,000 µM rolipram.
Thalidomide analogs inhibit the LPS-induced pro-inflammatory TNF-α and IL-1β but increase the anti-inflammatory IL-10 in pregnant rat uterine explants
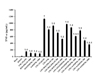
To evaluate the immunomodulatory and the anti-inflammatory properties of both thalidomide analogs, the preventive effects on production and release of the pro-inflammatory cytokines TNF-α (Fig. 5) and IL-1β (Fig. 6) were assessed in LPS-stimulated uterine explants of pregnant rats. The effects by forskolin and PDE-4 inhibitors on both pro-inflammatory cytokines showed a very similar pattern, because TNF-α and IL-1β were slightly, but significantly, elevated by forskolin (10 µM) or thalidomide analogs and rolipram at the highest concentration of 1000 µM in contrast to the basal concentration, which was not modified by DMSO. However, such rise in cytokine levels was quite distant from the several-fold increase induced by LPS; conversely, forskolin partially prevented that increase as well as the PDE-4 inhibitors diminished the production and release of these pro-inflammatory cytokines in a significant concentration-dependent manner. The order of efficacy as anti-inflammatory agents was as follows: 4APDPMe was the most effective (p<0.05) > rolipram > 4NO2PDPMe.
On the other hand, just to confirm the immunomodulatory effects by these PDE-4 inhibitors, the promoting activity on the anti-inflammatory cytokine IL-10 (Fig. 7) was also evaluated; in this case, only the highest concentration of 1000 µM of the PDE-4 inhibitors was tried. Forskolin (10 µM), rolipram and thalidomide analogs induced per se a similar augmentation in the concentration of IL-10 when compared with basal control, which resembled those observed in the pro-inflammatory cytokines though in a higher extent. In contrast, LPS did not elevate the IL-10 concentration in a significant way; nevertheless, PDE-4 inhibitors caused a remarkable increase of IL-10, despite the LPS-induced limiting effect. Rolipram provoked the highest concentration of IL-10 (p<0.05)>4APDPMe>4NO2PDPMe, while forskolin just yielded a modest increase in the concentration of that anti-inflammatory cytokine.
DISCUSSION
This study shows the utero-relaxant effect of two thalidomide analogs on the phasic and tonic contractions induced by PGF2α, K+ and Ca2+, which were strongly related to the cAMP accumulation due to the PDE-4 inhibition. Some PDE-4-inhibitors have different affinity to inhibit distinct PDE-4 isoforms; however, apremilast, a selective PDE-4 and TNF-α inhibitor from the same family of chemical structure of the tested thalidomide analogs, has a similar affinity for all PDE-4 isoforms [27]. Thus, the present results strongly suggest the participation of the PDE-4 isoforms in the pregnant rat uterine contraction, as reported in human myometrium for other PDE-4 inhibitors [13] and for the thalidomide analogs herein tried [6]. The pharmacological relaxant properties of both thalidomide analogs were assessed on phasic and tonic contractions in pregnant rat uterus. Hence, to evaluate if the inhibition of the contractions was a direct consequence of the inactivation of receptor/voltage-operated Ca2+ channels or by the stimulation of AC, controls of nifedipine and forskolin were used for comparison, since both compounds inhibit the uterine contraction by blocking Ca2+ channels and by increasing cAMP, respectively [2829]. The results were consistent with the findings of a study using rolipram [30], a selective PDE-4 inhibitor, and forskolin, both induced an increase of cAMP by inhibition of PDE-4 in the near-term human myometrium. The thalidomide analogs inhibited the PGF2α-, K+- and Ca2+-induced contractions in a concentration-dependent manner, which indicates that both analogs have similar actions to rolipram in potency and efficacy, suggesting that both analogs may recognize the same isoforms of PDE-4s [1730]; then, although there were minor differences in potency of both analogs and rolipram, it is possible that they share the same targets in pregnant rats as they did in human pregnant myometrium [6].
Myometrial contraction is positively regulated by myosin light chain kinase (MLCK) and negatively by MLC phosphatase (MLCP). Ca2+-calmodulin activate MLCK whereas MLCP activation depends on cAMP presence [31]. It has been patently suggested that the mechanism of action of PDE-4 inhibitors to provoke the relaxation of uterine smooth muscle occurs via PKA-mediated phosphorylations, which may activate or block diverse protein targets, such as the voltage-operated calcium channels (VOC) or receptor-operated calcium channels (ROC) [6]. Since PDE-4-inhibitors increase intracellular cAMP concentrations and activate PKA, the tonic and phasic contractions are suppressed easily, suggesting the inhibition of other target proteins through PKA-mediated phosphorylation, such as Rho-kinase (ROCK), because ROCK phosphorylates the myosin phosphatase-targeting subunit (MLCP-MYPT1), diphosphorylates the myosin regulatory light chain, and phosphorylates MLCP to cause contraction, as reported in vascular smooth muscle and human myometrium [323334]. Further evidence indicates that some PDE-4 inhibitors could block the calcium influx, such as rolipram and the thalidomide analogs herein tested, because of their similar structure to nifedipine, verapamil and nicardipine, what leads to uterine relaxation [62935]; in fact, the present results again suggest that both analogs blocked the calcium influx. In sum, the results are in agreement with other reports where forskolin and rolipram [3637], as well as these thalidomide analogs cause uterine relaxation by increasing cAMP and perhaps by the novel mechanism of action of rolipram and thalidomide analogs as potential Ca2+-channel blockers [6].
LPS is a strong inducer of immune responses in premature parturition of either humans or animals [3839]. Both thalidomide analogs produced an inhibition on the LPS-induced release of TNFα and IL-1β, which was comparable to the effect of rolipram and other PDE-4 inhibitors as well as other thalidomide analogs on cytokine secretion from macrophages, monocytes and peripheral blood mononuclear cells [184041]. The proposed signaling pathways involved in the inhibition on TNFα are via cAMP/PKA and NF-κB inhibition [4243]. The inactivation of NF-κB transcription [4445] did not affect the IL-10 secretion [46], it was actually increased, because IL-10 is transcribed by seven different transcription factors in macrophages [47] and because the IL-10 promoter has four cAMP response element targets [46]. The class of thalidomide analogs herein proved have not only been reported as utero-relaxing agents, but also have been evaluated as hepatoprotectors because of their immunomodulatory effects, which are capable of increasing IL-10 [62448].
The search for new drugs for the treatment of preterm labor tries to identify target molecules involved in the signaling pathways that lead to uterine contractions. As commented, salbutamol or terbutaline are, without any significant clinical benefit, commonly used as tocolytic agents, but are often associated with severe fetal and maternal side effects [1516]. The down-regulation of proinflammatory cytokines synthesis and the increase of cAMP through the combination of tocolytics and PDE-4 inhibitors may dampen the inflammatory cascade, which leads to premature contractions [1749]. Recent clinical trials with apremilast, one of main thalidomide analogs designed as PDE-4-inhibitor for inflammatory disorders, has been approved for the treatment of autoimmune diseases, such as psoriasis and psoriatic arthritis [50]. The increase of cAMP and the decrease of pro-inflammatory cytokines, suggest the thalidomide analogs as potentially useful for the treatment of preterm labor, when the molecular target is PDE-4, particularly the PDE-4B isoforms [6751], as well as for the treatment of autoimmune ailments. Further studies are required to determine in detail the interactions of both analogs with different PDE-4 isoforms; nevertheless, the utero-relaxant and anti-inflammatory effects of the thalidomide analogs were associated with the increased cAMP levels as PDE-4 inhibitors in the pregnant rat uterus.
Concerning the possible side effects of these non-teratogenic thalidomide analogs, the reports indicate that they appear to be non-toxic and non-mutagenic when they are tried in cell culture or in acute and chronic animal models [6182448]. Probably, these thalidomide analogs may have similar side effects than apremilast which has an acceptable safety profile; indeed, the observed side effects in patients are mild or moderate gastritis, diarrhea and nausea with a tendency for resolution [52]. Moreover, rolipram shows the same side effects but they are severe. In conclusion, the immunomodulatory, anti-inflammatory, and uterus-relaxant properties of 4APDPMe and 4NO2PDPMe, as PDE-4 inhibitors and probable Ca2+-channel blockers, place them as potentially safe and effective tocolytic agents in a field that urgently needs improved pharmacological treatments, as in cases of preterm labor.






 XML Download
XML Download