

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The subnucleus caudalis of the spinal trigeminal sensory nucleus is frequently termed the medullary dorsal horn (MDH) owing to its anatomical and physiological similarity with the spinal dorsal horn. The small myelinated Aδ-fiber and unmyelinated C-fiber enter the spinal trigeminal tract and terminate primarily in lamina I and II of the MDH. Therefore, the MDH is known as the center for processing pain from the craniofacial area, and for transmitting the integrated signals to the upper brain [12].
Reactive oxygen species (ROS), such as superoxide anion (O2·−), hydrogen peroxide (H2O2), and nitric oxide (NO) contribute to the increase of pain hypersensitivity during persistent pain [34]. Recently, Wang et al. reported that application of superoxide dismutase (SOD), which breaks down O2·−, prevents the development of inflammation and hyperalgesia following the injection of an inflammatory agent into the paw [5]. Other studies have reported an increased ROS production in the dorsal horn following spinal cord injury (SCI), which was attenuated by treatment with ROS scavengers such as phenyl N-tertbutylnitrone (PBN) or 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL) [67]. Furthermore, ROS accumulation was observed primarily in the mitochondria of dorsal horn neurons after capsaicin treatment [8]. These results suggest that O2·− participates in the nociceptive signaling cascade.
Increased production of ROS has been shown to affect membrane excitability related to the activity of ion channels. For example, voltage-dependent Na+, K+, and Ca2+ channels, Ca2+-activated K+ channels, and KATP channels have been identified as targets for ROS [910]. In addition, H2O2 induced membrane depolarization and increased excitability in medium spiny neurons via activation of TRP channels [11]. Moreover, NO, induced by an application of sodium nitroprusside, have been reported to influence the membrane potential of spinal substantia gelatinosa neurons via modification of various K+ channels and nonspecific cation channels (NSCC) [12].
Xanthine oxidase (XO) is an important enzymatic source of O2·−. This enzyme catalyzes the conversion of hypoxanthine and xanthine to uric acid, which produces an enormous amount of O2·− [13]. XO levels have been reported to increase in a model of neuropathic pain [14], and increased XO activity and O2·− production has been suggested to play a role in the development of pain [15]. However, the effects of O2·− generated by XO on the excitability of MDH neurons and the underlying ionic mechanisms have not been elucidated.
In this study, we investigated whether ROS derived from XO contribute to pain transmission by affecting the membrane potential and the ionic current of MDH neurons. For that, we used patch-clamp recordings from transverse slices of the spinal trigeminal nucleus. To our knowledge, this is the first study in which the effects of O2·− generated by the X/XO system on membrane excitability in MDH neurons were investigated.
METHODS
Brain slice preparation
Sprague-Dawley rats (13~18 days old) were anesthetized with ether. The procedures were approved by the University of Wonkwang Committee on Ethics in the Care and Use of Laboratory Animals (WKU09-076). Rats were decapitated and the brain stem was rapidly removed and placed in an ice-cold solution at 0~4℃. The brain stem was glued to the stage of a vibrating microslicer (752M, Campden Instruments, Loughborough, UK) and cut into 150~350 µm-thick slices. Slices were subsequently incubated at 32℃ for at least 1 h with 95% O2 and 5% CO2. Then, slices were transferred to a recording chamber mounted on an upright microscope.
Solutions and drugs
The dissecting solution used for the preparation of brain slices was composed of: sucrose (252 mM), KCl (2.5 mM), CaCl2 (0.1 mM), MgCl2 (2 mM), Glucose (10 mM), NaHCO3 (26 mM), and NaH2PO4 (1.25 mM). The extracellular fluid used for the patch-clamp recording contained: NaCl (117 mM), KCl (3.6 mM), CaCl2 (2.5 mM), MgCl2 (1.2 mM), NaH2PO4 (1.2 mM), NaHCO3 (25 mM), and glucose (11 mM). It was continually aerated with 95% O2 and 5% CO2, which maintained the pH at approximately 7.4. The pipette (internal) solution contained: K-Glu (150 mM), HEPES (10 mM), KCl (5 mM), EGTA (0.1 mM), Mg-ATP (2 mM), and NaGTP (0.3 mM). The pH was adjusted to 7.2 using KOH. In low Na+ external solution, equimolar choline-Cl replaced NaCl. The following reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA): X, XO, PBN, catalase, superoxide dismutase (SOD), 4,4-diisothiocyanatostilbene-2,2-disulfonic acid (DIDS). Dihydroethidium (DHE) and 2′,7′-dichlorofluorescin diacetate (DCF-DA) were obtained from Molecular Probes (Eugene, OR, USA).
Patch-clamp recording
Microelectrodes were prepared from capillary glass tubes (TW150-3, WPI, USA) using a microelectrode pipette puller (PP830, Narishige, Japan). Patch pipettes, filled with the pipette solutions, were used at a resistance ranging from 6 to 8 MΩ. The MDH of the spinal trigeminal nucleus was viewed using an upright microscope (BX50WI, Olympus, Japan). Membrane potentials and currents were recorded using an Axopatch 200B amplifier (Axon Instruments, USA) that was connected to a computer using an A/D converter (Digidata 1322A, Axon Instruments, USA). Membrane potential recording and data analyses were performed using the pClamp software (version 9.0, Axon Instruments, USA). Generated currents were filtered with a low-pass 8-pole Bessel filter at 2 kHz. All experiments were performed at room temperature (22±1℃).
Fluorescence imaging
The slices were loaded with fluorescence dyes including DCF-DA and DHE for 10~30 min at 30℃. The slices were examined on an inverted fluorescence microscope (x200; LSM 510, Carl Zeiss, Germany). Confocal laser scanning microscopy was used with two lasers, i.e. an argon ion laser emitting at 488 nm and a HeNe laser emitting at 543 nm to excite FITC and Cy3, respectively. A time series was used to record images every 30 s. The fluorescence intensity was analyzed in predefined a region of interest (ROI).
RESULTS
Effects of X/XO on the membrane excitability in MDH neurons
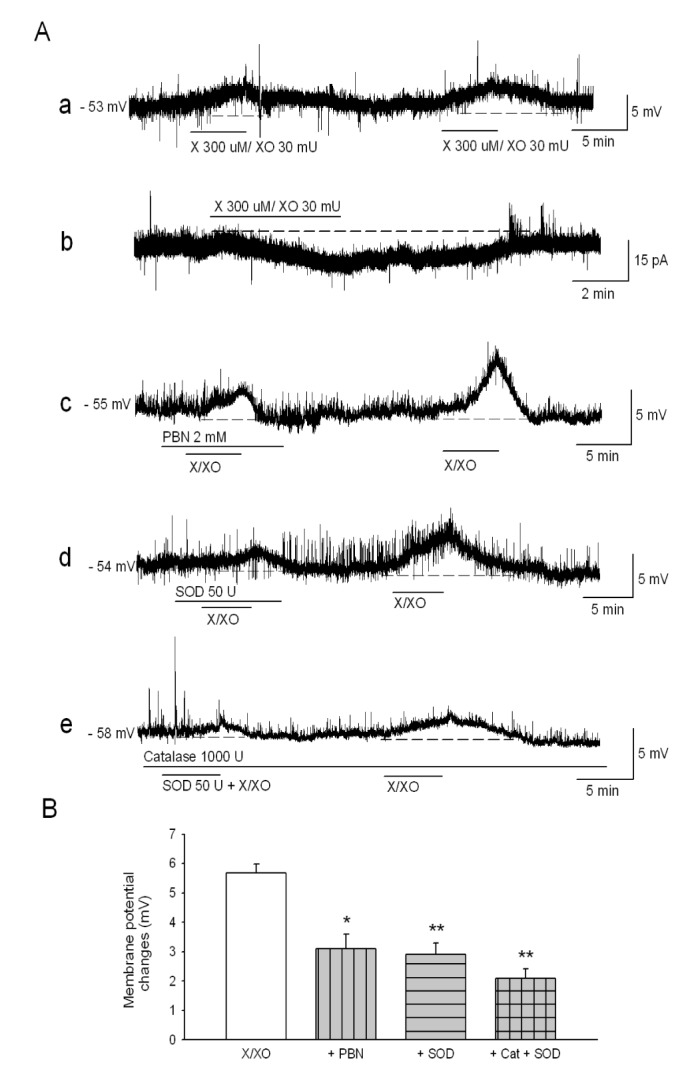
We examined whether the xanthine plus xanthine oxidase (X/XO) affects membrane excitability in MDH neurons. During current-clamp recording, the administration of X/XO (300 µM/30 mU) for 5 min induced a reversible membrane depolarization (5.6±0.3 mV, n=95). When an MDH neuron was clamped at a holding potential of −60 mV in voltage clamp recording, X/XO induced an inward current (−8.2±0.6 pA, n=20). To determine whether X/XO-induced changes were due to the release of ROS, MDH neurons were pretreated with ROS scavengers, including PBN (2 mM), SOD (50 U), and catalase (1000 U) plus SOD, prior to the application of X/XO. X/XO-induced depolarization significantly reduced in the presence of the ROS scavengers, such as PBN (3.1±0.5 mV, n=7, p<0.05), SOD (2.9±0.4 mV, n=7, p<0.01), and catalase plus SOD (2.1±0.3 mV, n=10, p<0.01) (Fig. 1). These results suggest that ROS are released by X/XO, which in turn induces changes in the membrane excitability of MDH neurons.
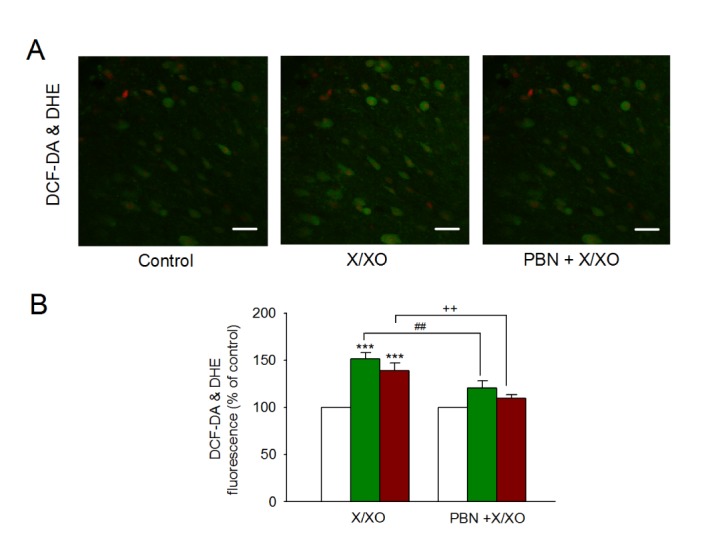
Furthermore, intracellular ROS generation by X/XO was assessed using DCF-DA and DHE. The intracellular fluorescence intensity in DCF-DA- and DHE-loaded cells increased during perfusion of X/XO for 5 min (151.8±6.2%, n=8, p<0.001, and 139.2±7.9%, n=8, p<0.001), which was inhibited by the ROS scavenger PBN (120.6±7.5%, n=5, p<0.01, and 110.0±3.4%, n=5, p<0.01) (Fig. 2).
The ionic mechanism underlying X/XO-induced responses
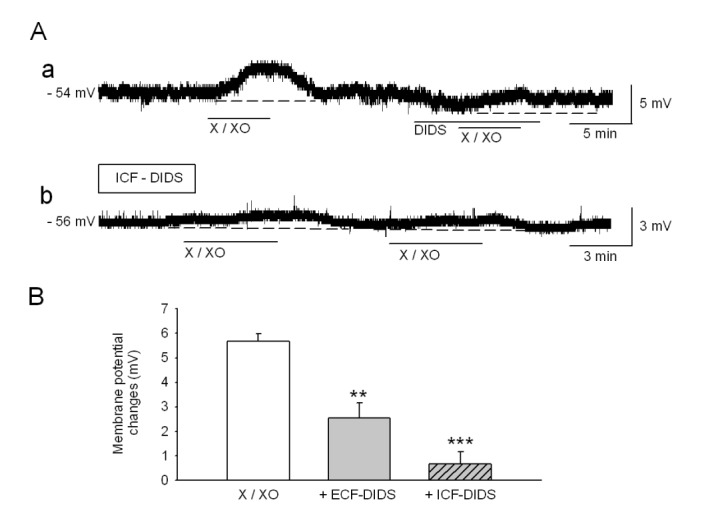
The addition of X/XO is known to initially generate O2·−, which has a low permeability in the membrane [16]. Moreover, extracellular O2·− can enter the cell through the anion channel and can participate in intracellular signaling either directly or by binding to other substances [17]. To confirm whether indeed X/XO-derived O2·− enters the cell via anion channels, we applied DIDS, an anion channel blocker. The application of DIDS (300 µM) within the bath (external) or the pipette (internal) solution significantly inhibited depolarization by X/XO (2.5±0.6 mV, n=11, p<0.01 and 0.6±0.5 mV, n=7, p<0.001, respectively) (Fig. 3). These results suggest that O2·− moves from the extracellular into the intracellular space through anion channels, and subsequently contributes to intracellular signaling.
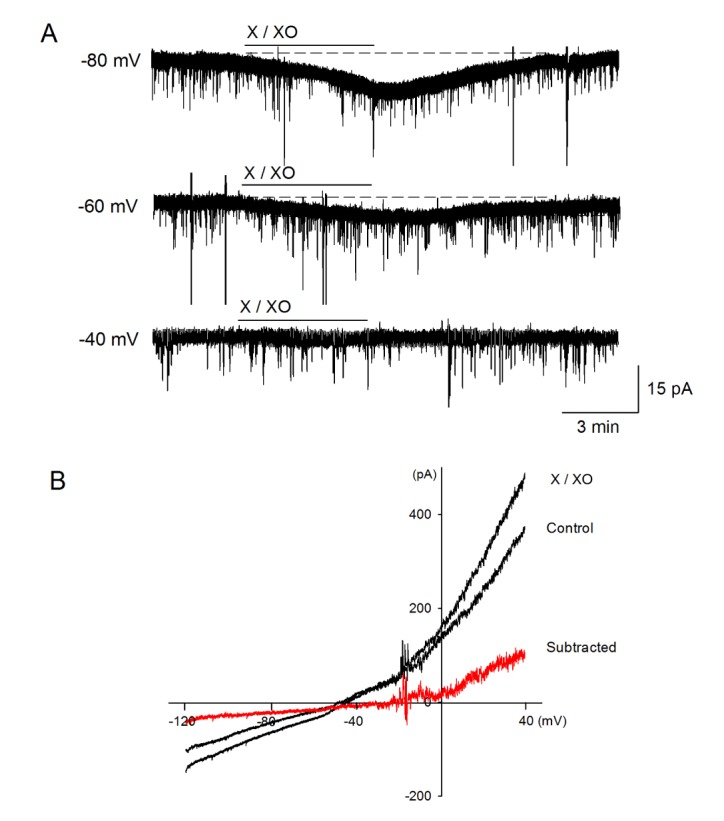
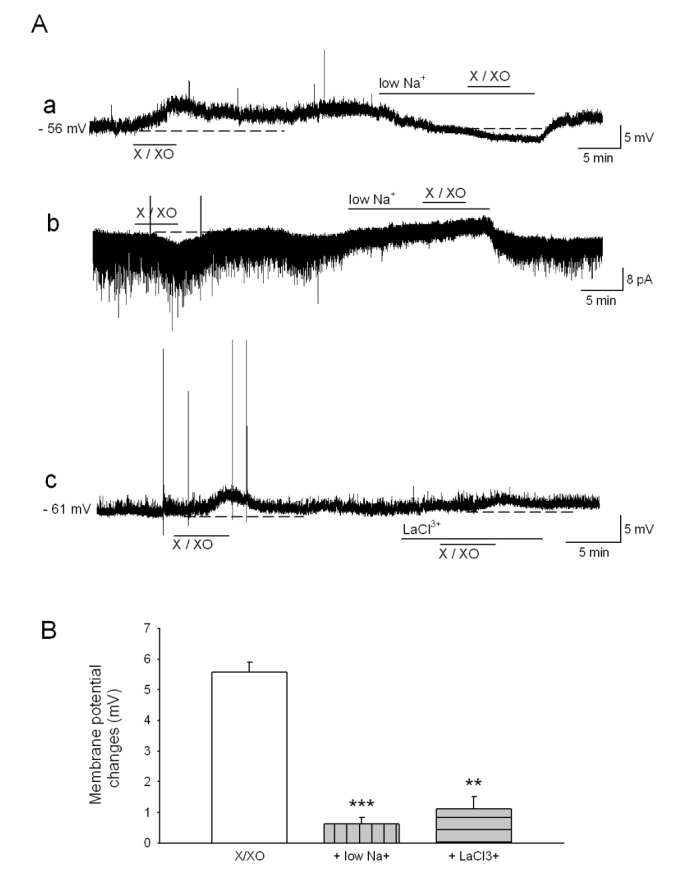
We next investigated which ions generate X/XO-induced depolarization and inward currents. In the voltage clamp condition, X/XO was applied at different holding potentials (–80 mV, –60 mV and –40 mV). X/XO-induced inward currents reduced at the depolarized holding potential. At a holding potential of –40 mV, no currents occurred. In addition, when a ramp pulse ranging from –120 to +40 mV was applied, the subtracted current (X/XO minus control) was reversed at a more positive than –40 mV (Fig. 4). Therefore, these findings suggest that the X/XO-induced current transports K+ as well as Na+. Furthermore, X/XO-induced depolarization is significantly reduced in the presence of the external low Na+ solution and an NSCC blocker, La3+ (100 µM) (0.6±0.3 mV, n=8, p<0.001 and 1.1±0.5 mV, n=13, p<0.01, respectively) (Fig. 5). These results indicate that membrane depolarization and inward currents are induced by influx of Na+ ions via NSCC.
DISCUSSION
ROS are possibly involved in the pathophysiology of diabetes, cancer, neurodegenerative diseases, and aging [181920]. However, recent studies have shown that physiological concentrations of ROS mediate reversible regulatory processes and signaling [21]. The contribution of ROS to nociceptive processing has been revealed by several studies. For example, ROS are produced into the spinal trigeminal nucleus during a persistent facial pain induced by a subcutaneous injection of formalin. Moreover, decreased superoxide dismutase (SOD) activity was associated with an increased pain behavior [22], suggesting that elevated ROS levels in the spinal trigeminal nucleus are sufficient to induce pain.
There are various sources to produce ROS during pain processing. For example, ROS can be generated from mitochondria (complexes I and III of the electron transport chain), xanthine oxidase, cyclooxygenases, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [23]. In this study, X/XO was applied in MDH neurons in order to induce a ROS generation. During the current-clamp recording, administration of X/XO for 5 min induced membrane depolarization, which was significantly reduced by ROS scavengers, such as PBN, SOD, and catalase (Fig. 1). In addition, ROS generation was confirmed by DCF-DA and DHE fluorescence imaging in slices containing MDH neurons (Fig. 2). These results suggest that ROS were produced following the application of X/XO, which in turn induced changes in membrane excitability of the MDH neurons.
Laser confocal microscopy has been used to investigate the role of ROS in cellular signaling, and several fluorescent proves have been designed to detect ROS that are associated with cellular oxidative stress. For example, DCF-DA is a relatively nonselective prove that reacts with free radical in the cytoplasm, nucleus, and mitochondria [24]. It diffuses passively into cells, where the acetate group is cleaved by intracellular esterase, and DCF remains inside the cells to develop fluorescence. DCF-DA is reported to detect not only H2O2, but also O2·−, hydroxyl radical (·OH), NO, and peroxynitrite (ONOO−) [25]. On the other hand, DHE prove is very sensitive to O2·−. When it meets O2·−, it is oxidized to the fluorescent product ethidium bromide. However, it has very low sensitivity to H2O2 and does not react well with ·OH or ONOO− [26].
The O2·− has a low permeability in the membrane [27]. Additionally, since O2·− is not only a free radical but also an anion, it is possible that its effect in the cell can occur by influx through the chloride channel [17]. Several previous studies have provided evidence for O2·− transport through anion channels, which could be effectively blocked by DIDS [2829]. Moreover, the DIDS is also known to block O2·− release from the mitochondria into the cytosol [30]. A voltage-dependent anion channel (VDAC) located in the mitochondrial outer membrane plays the role of a pore that releases mitochondria-generated O2·− to the cytosol [31]. In the present study, pretreatment of DIDS within both bath and pipette solution significantly inhibited X/XO-induced depolarization (Fig. 3). These findings indicate that X/XO-induced O2·− transfer from the extracellular into the intracellular space via anion channels. Therefore, O2·− contributes to the signaling intracellular pathway.
Depolarization of the plasma membrane was observed on the addition of oxidizing agents such as H2O2, t-BHP, or nitric oxide. This effect can be associated with activation of a NSCC, which is characterized by a linear I-V relationship [1232]. In our experiments, X/XO elicited an inward current associated with a voltage-independent linear current-voltage relationship that reversed near −40 mV (Fig. 4). Hung and Magoski [33] reported that the approximately −40 mV reversal potential suggests the involvement of voltage-independent NSCC. A reversal potential between −45 and +20 mV is dependent upon a varying degree of cation selectivity [34]. Moreover, X/XO-induced depolarization significantly reduced in the presence of low Na+ solution and La3+, an NSCC blocker (Fig. 5). These results indicate that X/XO-induced membrane depolarization and inward currents are induced by influx of Na+ ions through the NSCC.
In summary, the present study presents evidence that O2·− flux across the plasma membrane occurs through chloride channels. Moreover, membrane depolarization and inward current are induced by influx of Na+ ions via NSCC. These are essential steps for modulating the membrane excitability by O2·−, which generated by the X/XO system in spinal trigeminal MDH neurons. In conclusion, these results suggest that O2·−, in addition to its role as a toxin, can also be considered a signaling molecule for pain transmission.
 XML Download
XML Download