

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammation is a response of the innate immune system to invasion of microorganisms and is characterized by pain, heat, swelling, redness, and loss of function [1]. Although inflammation is a host defense mechanism, chronic inflammation is considered to be a leading cause of a variety of inflammatory/autoimmune diseases, such as rheumatoid arthritis, atherosclerosis, diabetes, sepsis, and even cancer [2]. Innate immune cells, such as macrophages, are primarily in charge of initiating inflammatory responses via recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs). Binding of pathogen particles to their cognate receptors results in activation of macrophages. Activated macrophages initiate inflammatory responses by producing inflammatory mediators, including reactive oxygen/nitrogen species (e.g., nitric oxide (NO)), prostaglandin E2 (PGE2)) and pro-inflammatory cytokines (e.g., tumor necrosis factor (TNF)-α, interleukin-1β (IL-1β), and IL-6) [345]. Molecular mechanisms of inflammatory responses in macrophages have been well characterized. Once macrophages initiate inflammatory responses, intracellular signaling pathways are immediately activated by induction of intracellular signaling cascades consisting of a variety of kinases, including Src and spleen tyrosine kinase (Syk) and mitogenactivated protein kinases (MAPKs) (e.g., extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinase (JNK), and p38), resulting in the activation of transcription factors such as nuclear factor-kappa B (NF-κB) and activator protein (AP)-1 [6789]. The activation of these transcription factors finally induces the expression and production of inflammatory mediators and pro-inflammatory cytokines by macrophages, which can result in inflammatory lesions that cause serious tissue damage. Therefore, intracellular signaling molecules produced during inflammatory responses are potential pharmaceutical targets to prevent and treat inflammatory diseases. Researchers have therefore actively focused on identifying and validating novel anti-inflammatory natural products such as saponins, flavonoids, and anthraquinones as well as chemical agents such as (E)-3-(3-methoxyphenyl)-1-(2-pyrrolyl)-2-propenone that specifically target intracellular signaling molecules in macrophages [101112131415].
Hydroquinone (benzene-1,4-diol; HQ) is a benzene-derived phenol that is found in industrial chemicals, cigarette smoke, byproducts of petroleum [16], and various kinds of foods and beverages [17]. HQ has been reported to have an inflammatory effect by reducing the expression of pro-inf lammatory cytokines, including TNF-α, IL-1β, IL-2, IL-6, and IL-10, through suppression of Akt kinase in the NF-κB pathway in LPS-stimulated macrophages [1819202122]. Moreover, HQ has been reported to modulate immune reactions by suppressing T cell proliferation [23] and promoting the release of IFN-γ and IL-4 by CD4+ T cells [2425]. In spite of these anti-inflammatory and immunomodulatory activities, HQ is a toxic benzene metabolite; there is therefore great interest in developing novel HQ derivatives that have potent anti-inflammatory and immunomodulatory activities with minimal cytotoxicity.
In this study, we explored the cellular and molecular mechanisms underlying the anti-inflammatory activity of JS-III-49, a novel HQ derivative, in macrophage-mediated inflammatory responses. Our results indicate that JS-III-49 exerts anti-inflammatory activity in LPS-stimulated macrophages by targeting Akt kinase and p38 MAPK in the NF-κB and AP-1 signaling pathways, respectively.
Go to : 
Methods
Materials
JS-III-49 (Fig. 1A) was provided by Professor Sanghee Kim (Seoul National University, Seoul, Korea). RAW264.7 cells and HEK293 cells were purchased from the ATCC (Rockville, MD, USA). Lipopolysaccharide (LPS, E. coli 0111:B4), N-nitro-L-arginine methyl ester hydrochloride (L-NAME), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), indomethacin, and polyethylenimine (PEI) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Roswell Park Memorial Institute (RPMI) 1640, Dulbecco's Modified Eagle's medium (DMEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS), streptomycin, penicillin, and L-glutamine were purchased from Gibco (Grand Island, NY, USA). Primers used for semi-quantitative reverse-transcriptase polymerase chain reaction (RT-PCR) and quantitative real-time PCR specific for cyclooxygenase (COX)-2, inducible nitric oxide synthetase (iNOS), interleukin (IL)-6, IL-1β, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were synthesized by and PCR premix was purchased from Bioneer Incorporated (Daejeon, Korea). Antibodies specific for total or phosphorylated IκBα, p65, p50, Src, Syk, Akt, c-Jun, C-Fos, ERK, JNK, p38, LaminA/C, and α-actin were purchased from Cell Signaling Technology (Beverly, MA, USA). TRI reagent® was purchased from Molecular Research Center Incorporated (Cincinnati, OH, USA). Lipofectamine® 2000 reagent was purchased from Thermo Fisher Scientific (Waltham, MA, USA), and an enhanced chemiluminescence system was purchased from AbFrontier (Seoul, Korea). The enzyme immunoassay (EIA) kits used to determine PGE2 level were purchased from Amersham (Little Chalfont, Buckinghamshire, UK).
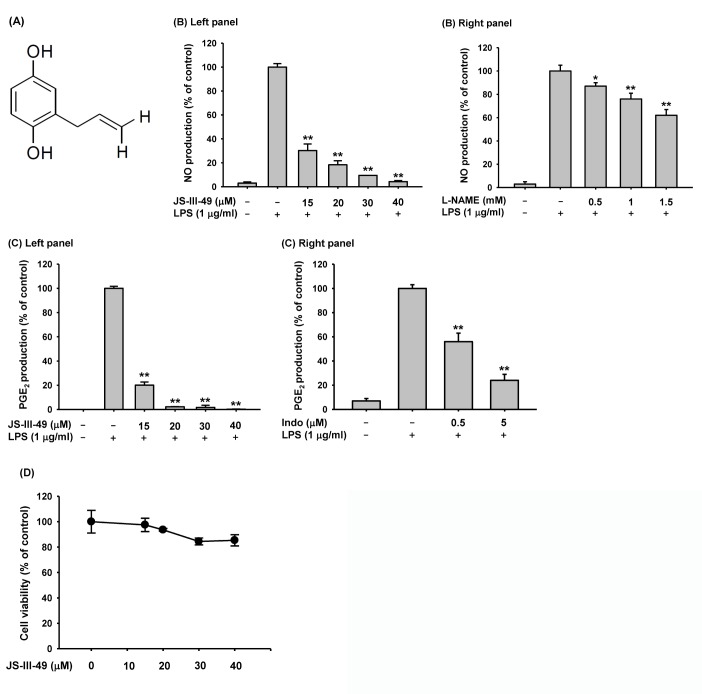 | Fig. 1Suppressive effect of JS-III-49 on the production of inflammatory mediators.(A) Chemical structure of JS-III-49. (B) RAW264.7 cells pretreated with the indicated doses of (left panel) JS-III-49 or (right panel) L-NAME for 30 min were treated with LPS (1 µg/ml) for 24 h, and NO levels in the cell culture media were determined. (C) RAW264.7 cells pretreated with the indicated dose of (left panel) JS-III-49 or (right panel) indomethacin (Indo) for 30 min were treated with LPS (1 µg/ml) for 24 h, and PGE2 levels in the cell culture medium were determined by EIA. (D) RAW264.7 cells were treated with the indicated dose of JS-III-49 for 24 h, and the cell viability was measured by MTT assay. *p<0.05, **p<0.01 compared to control.
|
Cell culture
RAW264.7 cells and HEK293 cells were cultured in RPMI 1640 medium and DMEM, respectively, each containing 10% heat-inactivated FBS, 100 mg/ml streptomycin, 100 U/ml penicillin, and 2 mM L-glutamine at 37℃ in a 5% CO2 humidified incubator. Cells were split and refreshed with fresh medium three time a week.
NO production assay
RAW264.7 cells (1×106 cells/ml) pretreated with either JS-III-49 or L-NAME at the indicated concentrations for 30 min were incubated with LPS (1 µg/ml) for 24 h. The NO production level was determined using Griess reagent as described previously [26].
PGE2 production assay
RAW264.7 cells (1×106 cells/ml) pretreated with either JS-III-49 or indomethacin at the indicated concentrations for 30 min were incubated with LPS (1 µg/ml) for 24 h. Production and the release of PGE2 in the cell culture medium were determined by enzyme immunoassay (EIA) as described previously [27].
Cell viability assay
RAW264.7 cells (1×106 cells/ml) were treated with JS-III-49 for 24 h, and the cytotoxic effect of JS-III-49 was determined by MTT assay as reported previously [2829]. Briefly, 100 µl of cell culture medium was mixed with 10 µl of MTT solution (10 mg/ml in phosphate-buffered saline (PBS), pH 7.4), and 15% of sodium dodecyl sulphate solution was directly added to the mixture after 4 h to stop the reaction, followed by further incubation for 24 h. The OD was measured at 570 nm using a Spectramax 250 microplate reader.
Semi-quantitative RT-PCR and quantitative real-time PCR
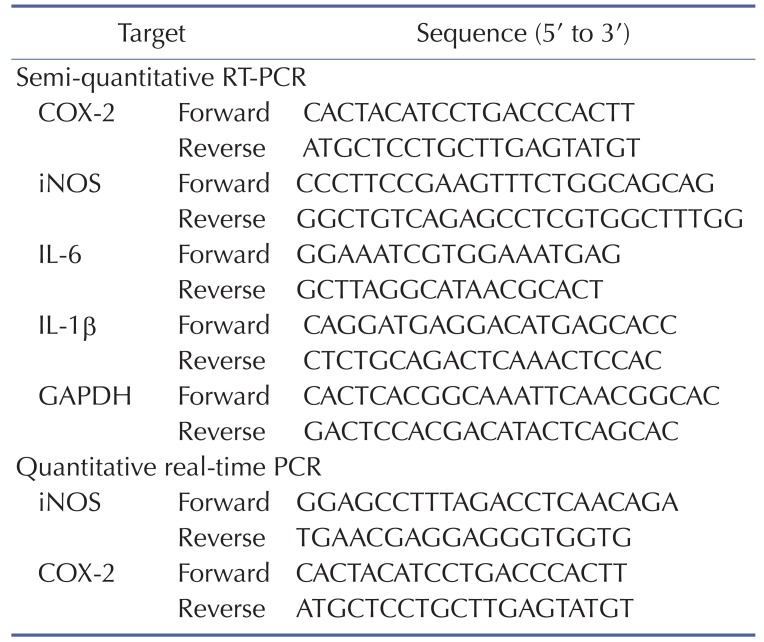
To determine mRNA expression levels of COX-2, iNOS, IL-6, and IL-1β, total RNA was extracted from RAW264.7 cells pretreated with JS-III-49 (0~30 µ?) for 30 min before incubation with LPS (1 µg/ml) for 6 h using TRI reagent® according to the manufacturer's instructions, and the total RNA was immediately used for cDNA synthesis. cDNA was synthesized from 1 µg of total RNA using MuLV reverse transcriptase according to the manufacturer's instructions, and semi-quantitative RT-PCR and quantitative real-time PCR were conducted as described previously [3031]. The sequences of the primers used for PCR are provided in Table 1.
Preparation of total/nuclear lysates and Western blot analysis
RAW264.7 cells were washed three times with PBS and lysed in ice-cold lysis buffer [32] using a sonicator (Thermo Fisher Scientific, Waltham, MA, USA) for 1 min at 4℃. Clear total lysates were separated from the pellet by centrifugation at 16,000 ×g for 10 min at 4℃ and stored immediately at –80℃ until use. Nuclear lysates of RAW264.7 cells were prepared as described previously [33] and stored immediately at –80℃ until use. For Western blot analysis, total and nuclear lysates were subjected to SDS-polyacrylamide electrophoresis and then transferred to polyvinylidene fluoride membranes. Total and phosphorylated forms of the target proteins were detected by antibodies specific for each target and then visualized by enhanced chemiluminescence according to the manufacturer's instructions.
In vitro kinase assay
The in vitro effects of JS-III-49 on the activities of purified targets, such as Src and Syk, were determined using the kinase profiler service of Millipore (Billerica, MA, USA) as reported previously [34]. Briefly, each purified target protein was incubated with 25 µl of reaction buffer containing MgATP for 40 min at room temperature, followed by incubation with 3% phosphoric acid solution to stop the reaction. Ten microliters of each incubate was dropped onto a P30 filtermat, which was washed three times with phosphoric acid for 5 min and one time with methanol for 5 min prior to drying and scintillation counting.
Statistical analysis
All data presented in this study are expressed as mean and standard deviation (SD) of at least three independent replicates. To compare statistical significance between groups, all results were analyzed by ANOVA/Scheffe's post hoc test and the Kruskal-Wallis/Mann-Whitney test. p<0.05 was considered to be statistically significant, and all statistical analyses were performed using SPSS (SPSS Inc., Chicago, IL, USA).
Go to :
Results and Discussion
HQ, a hydroxylated benzene derivative, has previously been reported to have immunosuppressive and anti-inflammatory functions in immune cells, especially macrophages [203536]. Therefore, many efforts have been made to synthesize and evaluate new HQ derivatives to find those with good immunosuppressive and anti-inflammatory efficacy but lower toxicity than HQ. In this study, we investigated the anti-inflammatory function of JS-III-49, a novel HQ derivative, in the macrophage-mediated inflammatory response. First, we examined the effect of JS-III-49 on the production of inflammatory mediators in macrophages by evaluating the production of NO and PGF2 in murine macrophage-like cells (RAW264.7 cells). As shown in Fig. 1B (left panel), JS-III-49 significantly suppressed the production of NO in LPS-stimulated RAW264.7 cells in a dose-dependent manner. As a positive control, the suppressive effect of L-NAME, a water-soluble inhibitor of NOS, on the production of NO was also examined in LPS-stimulated RAW264.7 cells [37]. As expected, L-NAME suppressed the production of NO in a dose-dependent manner in LPS-stimulated RAW264.7 cells (Fig. 1B right panel). Interestingly, JS-III-49 had a greater suppressive effect on NO production at much lower doses than L-NAME, suggesting that JS-III-49 has potent anti-inflammatory activity. JS-III-49 also suppressed the production of PGE2 in LPS-stimulated RAW264.7 cells in a dose-dependent manner (Fig. 1C left panel). Indomethacin, a non-steroidal anti-inflammatory drug (NSAID), was used as the positive control. Like JS-III-49, indomethacin suppressed the production of PGE2 in LPS-stimulated RAW264.7 cells in a dose-dependent manner (Fig. 1C right panel). Although both JS-III-49 and indomethacin showed anti-inflammatory activities by suppressing PGE2 production in macrophages, it is hard to directly compare their anti-inflammatory potency because of the dose ranges; further studies are required to compare the anti-inflammatory potency of JS-III-49 with that of indomethacin to evaluate whether JS-III-49 is more efficacious than this standard anti-inflammatory drug.
We next examined the cytotoxicity of JS-III-49 in RAW264.7 cells by MTT assay. Although JS-III-49 suppressed the production of NO and PGE2 at higher doses, such as 30 and 40 µ?, no significant cytotoxicity was observed at these doses of JS-III-49 (Fig. 1D), indicating that the suppressive effect of JS-III-49 on the production of NO and PGE2 was not a result of cell death.
iNOS and COX-2 are responsible for the production of NO and PGE2 in macrophages, respectively. Therefore, we investigated whether JS-III-49 suppressed the production of NO and PGE2 by down-regulating the mRNA expression of iNOS and COX-2 in RAW264.7 cells by semi-quantitative RT-PCR. JS-III-49 down-regulated the mRNA expression of both iNOS and COX-2 in LPS-stimulated Raw264.7 cells in a dose-dependent manner (Fig. 2A), strongly indicating that JS-III-49 suppresses the production of NO and PGE2 by down-regulating the expression of iNOS and COX-2 at the transcriptional level in macrophages. Suppressive effect of JS-III-49 on the expression of the pro-inflammatory cytokines IL-6 and IL-1β at the transcriptional level was also examined in RAW264.7 cells. Similar to the findings for iNOS and COX-2, JS-III-49 down-regulated the mRNA expression of these genes in LPS-stimulated RAW264.7 cells in a dosedependent manner (Fig. 2A). The suppressive effect of JS-III-49 on the mRNA expression of iNOS and COX-2 was further examined in RAW264.7 cells by quantitative real-time PCR. In agreement with the semi-quantitative RT-PCR results, JS-III-49 significantly down-regulated the mRNA expression of both iNOS (Fig. 2B) and COX-2 (Fig. 2C) in LPS-stimulated RAW264.7 cells in a dose-dependent manner. These results suggest that JS-III-49 exerted its anti-inflammatory effects in macrophages by modulating the mRNA expression of inflammatory enzymes and pro-inflammatory cytokines, thereby suppressing the production of the inflammatory mediators NO and PGE2.
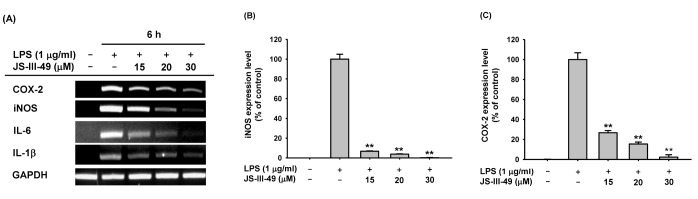 | Fig. 2Suppressive effect of JS-III-49 on the mRNA expression of pro-inflammatory genes.(A) RAW264.7 cells pretreated with the indicated doses of JS-III-49 for 30 min were treated with LPS (1 µg/ml) for 6 h, and mRNA expression levels of COX-2, iNOS, IL-6, IL-1β, and GAPDH were determined by semi-quantitative RT-PCR. (B, C) RAW264.7 cells pretreated with the indicated doses of JS-III-49 for 30 min were treated with LPS (1 µg/ml) for 6 h, and mRNA expression levels of (B) iNOS and (C) COX-2 were determined by quantitative real-time PCR. GAPDH was used as an internal control to normalize the mRNA expression levels of target genes. **p<0.01 compared with control.
|
It has been well established that mRNA expression of inflammatory mediators and pro-inflammatory cytokines during an inflammatory response in macrophages is tightly regulated by inflammatory transcription factors, such as NF-κB and AP-1 [6789]. Therefore, we hypothesized that down-regulation of mRNA expression of the inflammatory mediators, iNOS and COX-2, as well as the pro-inflammatory cytokines, IL-6 and IL-1β, by JS-III-49 (Fig. 2), was mediated by suppression of such transcription factors. To investigate this, we examined whether JS-III-49 could suppress the nuclear translocation of NF-κB transcription factors p65 and p50 in LPS-stimulated RAW264.7 cells by Western blot analysis. As shown in Fig. 3A, JS-III-49 effectively suppressed nuclear translocation of both p65 and p50 60 min and 30 min after LPS stimulation in RAW264.7 cells, respectively. JS-III-49 also suppressed the activity of IκBα, an intracellular signaling molecule upstream of NF-kB (Fig. 3B). These results support our hypothesis that JS-III-49 suppresses the production and mRNA expression of inflammatory mediators and pro-inflammatory cytokines by inhibiting the activity of IκBα and nuclear translocation of NF-κB transcription factors p65 and p50 during an inflammatory response in macrophages. It is well-known that many intracellular signaling molecules in the NF-kB pathway, including Src, Syk, and Akt, are activated in the macrophage-mediated inflammatory response [73839]. Because JS-III-49 inhibited the activation of NF-κB in LPS-stimulated RAW264.7 cells, we next investigated which intracellular molecules in the NF-κB signaling pathway were targeted by JS-III-49. Phosphorylated levels of Src, Syk, and Akt were determined in RAW264.7 cells after treatment of the cells with LPS for 2, 3, and 5 min. JS-III-49 suppressed the levels of p-Src at 3 min and p-Syk at 3 and 5 min, whereas the level of p-Akt was suppressed by JS-III-49 at 2, 3, and 5 min (Fig. 3C). This finding was unexpected because Src and Syk are upstream of Akt; JS-III-49-mediated suppression of Akt phosphorylation was observed from 2 min to 5 min, while phosphorylation of its upstream signaling molecules, Src and Syk, was not affected by JS-III-49 at 2 and 3 min, which suggests that JS-III-49 directly targets Akt but not Src and Syk during the inflammatory response in macrophages. To test this possibility, we performed an in vitro kinase assay using purified Src and Syk. As expected, in vitro kinase activities of Src and Syk were not suppressed by JS-III-49 (Fig. 3D), indicating that neither Src nor Syk is a direct target of JS-III-49 during the inflammatory response in macrophages. To examine whether Akt is a direct target of JS-III-49 during the inf lammatory response in macrophages, we assessed levels of phosphorylated Akt in LPS-stimulated RAW264.7 cells treated with different doses of JS-III-49. As shown in Fig. 3E, JS-III-49 suppressed the level of p-Akt in a dose-dependent manner, strongly indicating that Akt is a direct target of JS-III-49, and that JS-III-49 specifically suppresses the activation of Akt during the inflammatory response in macrophages.
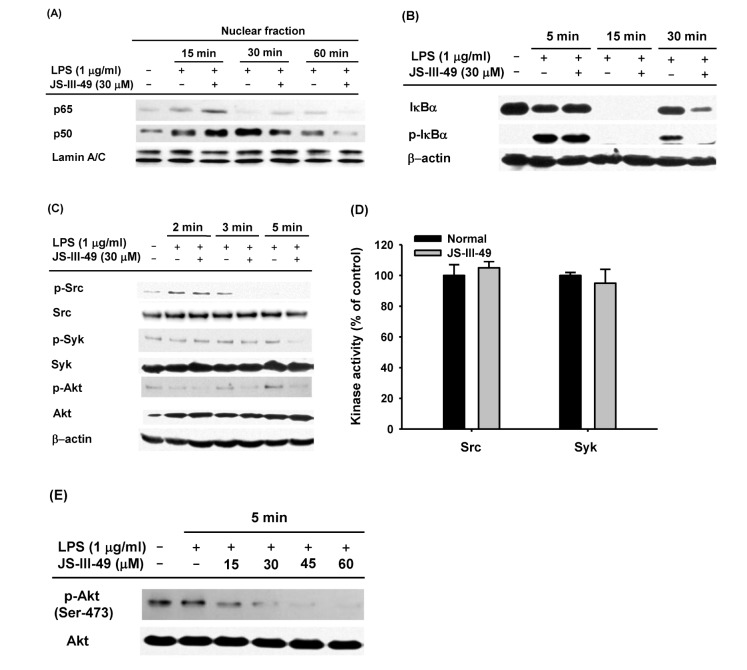 | Fig. 3The suppressive effect of JS-III-49 on the NF-κB pathway is mediated by direct targeting of Akt.(A) RAW264.7 cells pretreated with JS-III-49 (30 µM) for 30 min were treated with LPS (1 µg/ml) for the indicated time, and protein levels of p65 and p50 in the nuclear fraction of the cells were determined by Western blot analysis. (B) RAW264.7 cells pretreated with JS-III-49 (30 µM) for 30 min were treated with LPS (1 µg/ml) for the indicated time, and phosphorylated and total levels of IκBα in the total cell lysate were determined by Western blot analysis. (C) RAW264.7 cells pretreated with JS-III-49 (30 µM) for 30 min were treated with LPS (1 µg/ml) for the indicated time, and phosphorylated and total levels of Src, Syk, and Akt in the total cell lysate were determined by Western blot analysis. (D) Effects of JS-III-49 on the kinase activities of Src and Syk were determined by a conventional in vitro kinase assay using purified Src and Syk as described in the Materials and Methods. (E) RAW264.7 cells pretreated with the indicated doses of JS-III-49 for 30 min were treated with LPS (1 µg/ml) for 5 min, and phosphorylated and total levels of Akt in the total cell lysate were determined by Western blot analysis. Lamin A/C and β-actin were used as internal controls for the nuclear fraction and total cell lysate, respectively.
|
AP-1 is another critical transcription factor in the macrophage-mediated inflammatory response. Therefore, we examined whether JS-III-49 suppressed AP-1 during the inflammatory response in macrophages by examining the effect of JS-III-49 on the nuclear translocation of the AP-1 transcription factors c-Jun and c-Fos in LPS-stimulated RAW264.7 cells by Western blot analysis. As shown in Fig. 4A, JS-III-49 effectively suppressed the nuclear translocation of c-Fos 60 min after LPS stimulation in RAW264.7 cells, while it did not affect the nuclear translocation of c-Jun. Numerous previous studies have shown that mitogen-activated protein kinases (MAPKs) such as ERK, JNK, and p38 are upstream intracellular signaling molecules in the AP-1 pathway that are activated during the inflammatory response in macrophages [94041]. Because JS-III-49 suppressed the nuclear translocation of c-Fos in LPS-stimulated RAW264.7 cells, the suppressive effect of JS-III-49 on these MAPKs was next examined in LPS-stimulated RAW264.7 cells by Western blot analysis. Interestingly, JS-III-49 dramatically decreased the level of p-p38 at 30 min and 60 min after LPS stimulation of RAW264.7 cells, but had no effect on the phosphorylation of either ERK or JNK (Fig. 4B). These results strongly indicate that, during the inflammatory response in macrophages, JS-III-49 suppresses the activity of c-Fos by specifically inhibiting the activation of p38, which is an upstream MAPK in the AP-1 pathway.
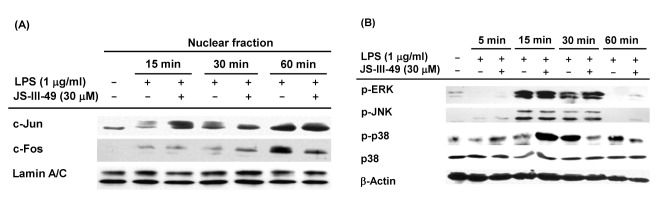 | Fig. 4The suppressive effect of JS-III-49 on the AP-1 pathway is mediated by targeting of c-Fos and p38.(A) RAW264.7 cells pretreated with JS-III-49 (30 µM) for 30 min were treated with LPS (1 µg/ml) for the indicated time, and protein levels of c-Jun and c-Fos in the nuclear fraction of cells were determined by Western blot analysis. (B) RAW264.7 cells pretreated with JS-III-49 (30 µM) for 30 min were treated with LPS (1 µg/ml) for the indicated time, and phosphorylated and total levels of ERK, JNK, and p38 in the total cell lysate were determined by Western blot analysis. Lamin A/C and β-actin were used as internal controls for the nuclear fraction and total cell lysate, respectively.
|
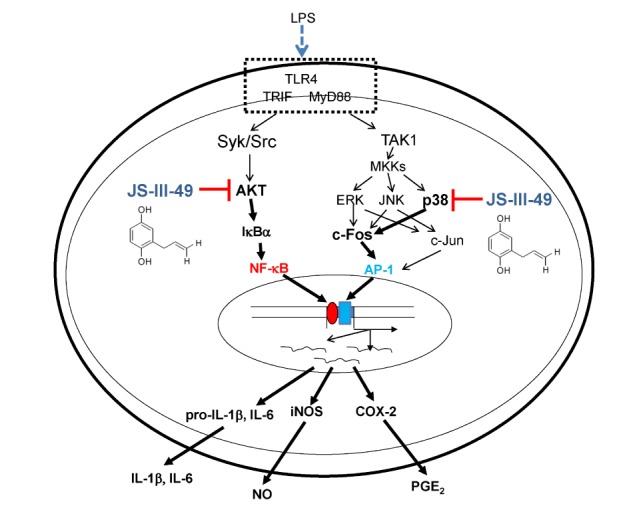
In conclusion, we explored the anti-inflammatory effects of JS-III-49, an HQ derivative, in macrophages, and demonstrated that JS-III-49 effectively suppressed the production and mRNA expression of inflammatory mediators as well as pro-inflammatory cytokines. This anti-inflammatory activity of JS-III-49 was achieved by direct inhibition of Akt, an upstream kinase in the NF-κB pathway, and p38, an upstream MAPK of c-Fos in the AP-1 pathway, as described in Fig. 5. These results suggest that JS-III-49 has the potential to prevent and treat inflammatory diseases by suppressing macrophage-mediated inflammatory responses.
Go to :
 XML Download
XML Download