

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dynamic changes of cytosolic Ca2+ level under physiological conditions are essential for cellular signalings of protein synthesis and signal transmission of neurons in central nervous system (CNS). However, due to its cytotoxic effects, excessive influx of Ca2+ and subsequent rapid elevation of [Ca2+]i can induce irreversible damage of neurons or result in pathological dysfunctions such as epileptic seizure. Hence, mammalian neurons exhibit several protective mechanisms to homeostatically regulate membrane excitability and cytosolic Ca2+ level. Several types of voltage dependent ion channels are involved in these neuroprotective processes via regulating cation flow. In its original description based on neuronal excitability, intrinsic excitability can be defined as the ability to fire action potentials (APs), which are determined by voltage-dependent K+ and Na+ channels (KV and NaV channels) [1]. Therefore, cation channels especially sensitive to membrane potential are key factors to regulate excitability in most neurons even though they are inhibitory. In particular, several subtypes of Kv channels can determine resting membrane potential and effectively induce cation outflow to downregulate neuronal excitability in many conditions of membrane depolarization [2].
Among the many types of K+ channels expressed in CNS neurons, Kv2.1 is a major component of delayed rectifier K+ channel (IDR channel), exhibiting sustained outward K+ currents [345]. This subtype plays a direct role in lowering membrane potential thus inhibiting AP initiation as well as limiting repetitive APs firing. Thus, the hyperpolarization of membrane potential and blockade of depolarization by Kv2.1 can prevent hyperexcitability that activates cytotoxic cascades and induces neuronal damage [67]. Previous studies showed that Kv2.1 channels are sensitive to changes in cytosolic Ca2+ level. For example, cytosolic Ca2+ increase and subsequent calcineurin activation affects the gating kinetics of IDR channels via dephosphorylating Kv2.1 channels, since calcineurin-dependent dephosphorylation of Kv2.1 decreases the threshold for IDR channel opening and disrupts channel clustering, resulting in changes of activation kinetics [18]. Kv2.1 channels have many serine, threonine, and tyrosine phosphorylation sites. These participate in the regulation of IDR channel kinetics; for e.g., PKA-mediated phosphorylation in serine/threonine sites induces the changes of gating kinetics but not membrane expression of Kv2.1 channels [910]. Dynamic changes of gating kinetics by de- or phosphorylation of Kv2.1 channels seem to be minor but significant for regulating K+ outflow and membrane excitability under physiological conditions. However, other studies have recently demonstrated that under several pathological conditions such as epileptic seizures and ischemic damages, acute increment of cytosolic Ca2+ rapidly potentiates IDR, and thus suppresses hyperexcitability [111213]. These results suggest that rapid changes of cytosolic Ca2+ may stimulate more effective signaling pathways by which IDR channels directly regulate membrane excitability. However, whether excessive Ca2+ influx rapidly and potentially enhances IDR remains unclear.
In the present study, we confirmed that dissociated hippocampal neurons of rats showed significant increase of IDR after high Ca2+ treatment, which could enhance synaptic activities and membrane excitability. The Ca2+-mediated IDR upregulation was sensitive to activity of voltage-dependent Ca2+ channels (VDCCs) but not NMDA receptors (NMDARs), suggesting a neuroprotective signaling pathway that is not targeted by postsynaptic Ca2+ influx via NMDARs. In the additional experiment, we confirmed that the contribution of Src family tyrosine kinase (SFK) was necessary for upregulating IDR under high Ca2+ condition, suggesting that the increased expression of IDR channels via SFK activation is a protective mechanism against excitotoxicity and pathogenic processes in neurons.
METHODS
Animal preparation and hippocampal primary culture
Hippocampal primary cultures were prepared from embryonic 20-day SD rats. After pregnant rats were deeply anesthetized, the embryos were removed and transferred to an ice-cold normal tyrode solution containing the following (in mM): 140 NaCl, 5.4 KCl, 2.3 MgCl2, 10 HEPES, 5 glucose. The pH level was adjusted to 7.4 with NaOH. Isolated hippocampi from embryonic rat brains were transferred to ice-cold minimal essential medium (MEM) containing Earle's salts and glutamine with 10% fetal bovine serum, 0.45% glucose, 1 mM sodium pyruvate, 25 µM glutamate and antibiotics, and the hippocampi were triturated. The cells were counted and seeded on glass coverslips coated with poly-L-lysine (Sigma-Aldrich) at a density of 9×104 cells/ml and transferred to an incubator gassed with 95% O2 and 5% CO2 at 37℃. After 7 hours, the whole plating medium was exchanged to Neurobasal medium (Sigma-Aldrich) containing B-27 (Invitrogen), and a half of the medium was replaced with new media once at DIV 5. Detail protocols for cell preparation and recording techniques were as previously reported [1415]. All experiments and procedures with animals were approved by the Animal Care and Use Committee of Jeju National University.
Drugs treatment
To electrophysiologically observe the changes of IDR in cultured hippocampal neurons, high Ca2+ condition was induced by doubling Ca2+ concentration of culture media (normal 1.8 mM) up to 3.6 mM (with CaCl2, Sigma-Aldrich) for 24 hours. APV (100 mM, Sigma-Aldrich) and nimodipine (10 mM, Sigma-Aldrich) were used with high Ca2+ treatment to block NMDARs and VDCCs, respectively, in some experiments. H89 was added to block the activation of PKA and ryanodine receptors and IP3 receptors of ER were blocked by Ryanodine and 2AP, respectively. Blockers of kinases and receptors were obtained from Sigma Aldrich. Caffeine or 4CMC (Sigma-Aldrich) were used to test the effects of CICR via ryanodine receptor activation. Details of drugs treatment are mentioned in results and legends of figures.
Electrophysiology
For patch-clamp recordings of dissociated hippocampal neurons, DIV 6~8 cultured cells were transferred to a recording chamber with a continuous flow of recording solution containing the following (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1.3 MgCl2, 10 HEPES, 10 glucose. pH level was adjusted to 7.4 with NaOH, and bubbled with 95% O2 and 5% CO2. TTX (0.5 µM, Tocrise) was added to the recording solution to block the Nav channels. The patch pipettes (4~6 MΩ) were filled with an internal solution containing the following (in mM): 20 KCl, 125 K+-gluconate, 4 NaCl, 10 HEPES, 0.5 EGTA, 4 ATP, 0.3 tris-GTP, and 10 phosphocreatin. pH and osmolarity were adjusted to 7.2 and 280~300 mOsm, respectively. In all experiments, neurons showing 7~11 pF whole cell capacitance were used. Whole cell recording parameters were monitored throughout each experiment and recordings where series resistance (6~20 MΩ) varied by more than 10% were rejected.
All recordings were performed at room temperature, low-pass filtered at 5 kHz, and digitized at 10 kHz by a Digidata 1322A convertor. Voltage-clamping whole cell patches, command pulse generation and data recording were performed using Axopatch 200B and pClamp 8 software. Thick-walled, filamented patch electrodes had tip resistance of 3~6 MOhm. For adjusting whole-cell parameters, membrane capacitance and series resistance were manually compensated in Axopatch 200B during applying command pulses. Series resistance varied between 8~30 MOhm, and recordings where series resistance varied by more than 10% were rejected. No electronic compensation for series resistance was employed during currents recording. Pulse commands to record sustained currents in voltage-clamping mode are described in detail in results and figures. Sustained K+ currents were digitally isolated using a prepulse protocol after subtracting leak currents. Peak currents were measured at +60 mV. All experimental data were additionally analyzed using IGOR pro software.
RESULTS
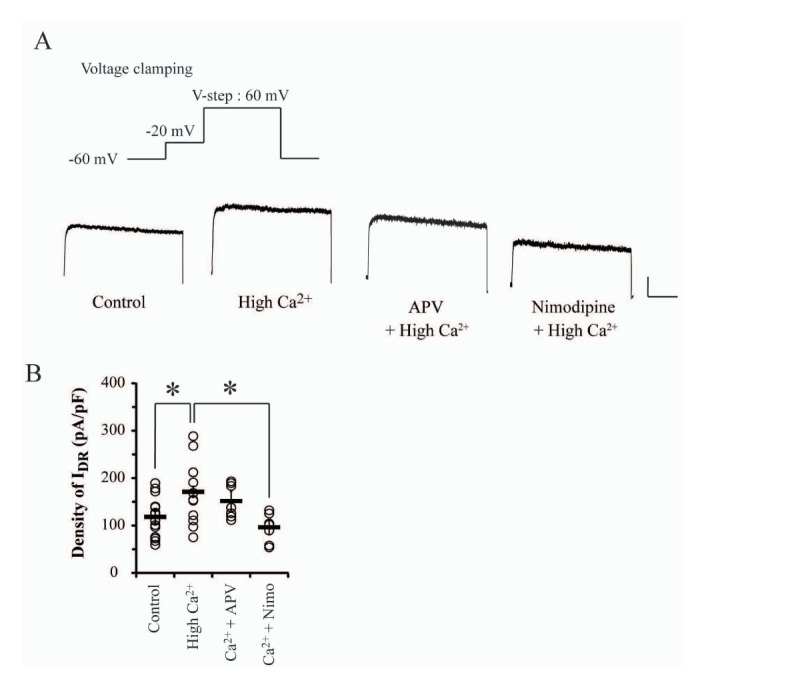
In physiological regulation of neuronal excitability, the dominant role of small but sustained K+ outflow through IDR channels is to determine and stabilize sub- and suprathreshold membrane potentials. However, signaling-mediated changes of its kinetics suggest a possibility that IDR channels may also be targeted by hyperexcitable conditions. In the present study, we tested K+ outflow through IDR channels in response to hyperexcitable condition induced by high Ca2+ overnight treatment (3.6 mM CaCl2, for 24 hours) in cultured hippocampal neurons. This experimental condition not only induces the downregulation of A-type K+ channels (IA channels) but also enhances synaptic Ca2+ influx by remodeling NMDARs composition, resulting in the hyperexcitability of hippocampal neurons [1416]. After treating high Ca2+ to culture media for 24 hours, neurons were transferred to a recording chamber and washed in normal ACSF solution for electrophysiological measurement. In results, the current density of IDR channels was significantly increased by high Ca2+ treatment, as shown in Fig. 1 (Control=112.62±10.28 pA/pF, n=13; high Ca2+=171.05±18.30 pA/pF, n=12, p=0.02). This indicates that IDR channels may be upregulated under hyperexcitable conditions.
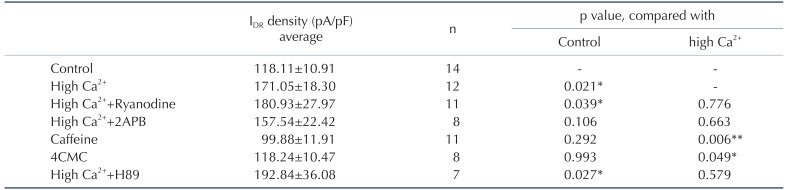
Depolarization of membrane potential activates two major Ca2+ influx pathways: VDCC and NMDAR. High Ca2+-enhanced neuronal excitability is reportedly dependent on Ca2+ influx via NMDARs, hence it was necessary to confirm if the increase of IDR was dependent on NMDARs [16]. APV (100 µM) or nimodipine (10 µM) with high Ca2+ treatment was applied to culture media for 24 hours before recording. APV-induced NMDARs block showed no effects on the high Ca2+-enhanced IDR in this experiment; whereas, nimodipine, a VDCCs antagonist, significantly inhibited the effect of high Ca2+ treatment on IDR (Fig. 1, APV=151.55±12.56 pA/pF, Nimodipine=96.03±9.34 pA/pF, p=0.48, 0.008 respectively, compared with high Ca2+). Thus, IDR upregulation may be triggered by Ca2+ influx through VDCCs rather than NMDARs. Furthermore, because it is well known that Ca2+-induced Ca2+ release (CICR) is responsive to Ca2+ influx, we also tested if it might participate in IDR upregulation resulted from Ca2+ influx via VDCCs. In Table 1, blocking of two major receptors of endoplasmic reticulum, ryanodine receptors (Ryanodine) and IP3 receptors (2APB), showed no effects on enhanced IDR under high Ca2+ condition. Additionally, caffeine or 4CMC, agonists of ryanodine receptors, showed no enhancement effects on IDR, unlike high Ca2+ treatment. These results strongly indicated that Ca2+ influx through VDCCs may directly regulate IDR channels regardless of CICR activation.
Although the current density of IDR was increased, the gating kinetics of IDR channels was not affected under high Ca2+ condition. In Fig. 2, the increase rate of IDR positively reflected the increase rates of command pulse potentials in all experimental groups. At +80 mV injection, significant differences of IDR amplitudes were observed between control and high Ca2+ treated neurons (Fig. 2A and B, Control=139.82±15.62 pA/pF; High Ca2+=226.79±17.34 pA/pF, p=0.02). The current density of IDR under high Ca2+ condition was enhanced up to 60% in each injection range from −40 to 80 mV compared with the control group. However, in the analysis of conductance compensated by each membrane potential, activation properties of IDR channels were consistent in all groups, indicating that the gating kinetics of IDR channels may not be subjected to high Ca2+ condition (Fig. 2C and D, Vh of activation: Control=12.38±2.62 mV; High Ca2+=22.21±4.4 mV, p=0.1). This result suggests that the upregulation of IDR channels under high Ca2+ condition may not be dependent on their dephosphorylation by calcineurin activated by Ca2+ signaling. Additionally, both antagonists, APV and nimodipine, showed no effects on the activation kinetics of IDR channels (Vh of activation; APV group=10.11±1.91; Nimodipine group=14.92±1.63, p=0.55 and 0.45, respectively).
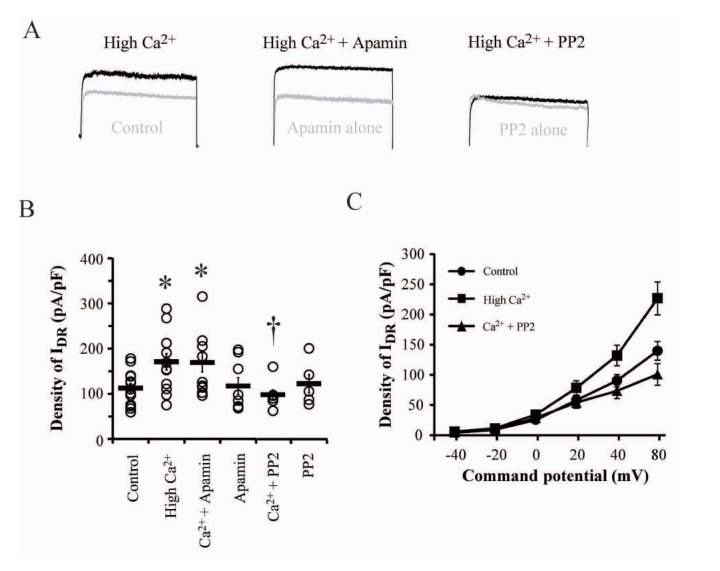
Previously, Ledoux et al. [17] reported that the enhancement of intracellular Ca2+ concentration induced the elevation of K+ efflux through small conductance Ca2+ channels (SK channels). Because of possible change in intracellular Ca2+ concentration under the experimental condition, it was necessary to confirm whether the enhanced density of IDR by high Ca2+ was contaminated by the flow of SK channels even though the two channels exhibit totally different gating mechanisms. In Fig. 3, the effect of 100 nM apamin with high Ca2+ was tested by adding to culture media for 24 hours to block SK channels. The addition of apamin did not influence the increased density of IDR by high Ca2+ treatment, indicating that upregulating IDR is not via the activation of SK channels (179.37±26.56 pA/pF, p=0.04 and 0.81, as compared with control and high Ca2+, respectively).
Ca2+ signaling cascades activated by Ca2+ influx through synaptic NMDARs induce the phosphorylation of protein kinase A (PKA) resulting in the downregulation of IA in hippocampal neurons [1518]. Although it is still under argument, it is possible that PKA regulates the turnover rate of proteins from plasma membrane and currents through various channels. In this study, PKA was not involved in the cellular processing to enhance IDR under high Ca2+ condition (Table 1, +H89). Rather, we confirmed that SFKs have a potentially important role in IDR upregulation under high Ca2+ condition (Fig. 3). Adding SFK blocker, PP2 (1 µM), to culture media with high Ca2+ showed no enhancement of IDR, with similar levels as in control neurons that were not treated with any drugs (Ca2++PP2=99.01±10.52 pA/pF, p=0.43 and 0.02 compared with the control and high Ca2+, respectively). Furthermore, PP2 alone showed no effect on the density of IDR, indicating that high Ca2+ condition possibly activate SFKs to prevent neuronal excitotoxicity or damage (123.46±20.23 pA/pF, p=0.63 compared with the control).
DISCUSSION
Neurons in CNS exhibit dynamic regulatory signalings to protect from hyperexcitable conditions that result in neuronal damage. Herein, we reported upregulation of IDR channels, as a neuroprotective regulator, which increased K+ outflow under hyperexcitable condition induced by high Ca2+ overnight treatment in cultured hippocampal neurons. The high Ca2+-induced enhancement of IDR was dependent on Ca2+ influx through VDCCs but not NMDARs and was not affected by CICRs that synergistically increase intracellular Ca2+ level (Fig. 1 and Table 1). These results indicate that hippocampal neurons may homeostatically modulate IDR channels in response to Ca2+ influx through VDCCs. Thus, neuronal excitability is consequently downregulated by the enhancement of K+ outflow via IDR channels. Furthermore, IDR enhancement was dependent on tyrosine kinases of Src family, indicating the possibility of an unique signaling cascade for the downregulation of excitability to prevent neuronal damages under hyperexcitable or pathogenic conditions.
High Ca2+ treatment for 24 hours used in this study (i.e. doubling Ca2+ concentration in culture media), is reported to mediate changes in the subunit composition of synaptic NMDARs, IA density and Ca2+-calmodulin dependent kinase activity [141619]. Increased Ca2+ influx through NMDARs and subsequent IA downregulation critically increases neuronal excitability due to changes in membrane resistance, firing rates of APs and after-hyperpolarizing potentials resulting from direct effects of the cationic flow through channels or receptors [1619]. These effects of high Ca2+ condition possibly result in irreversible damage to hippocampal neurons. However, hippocampal neurons showing Ca2+-induced downregulation of Kv4.2 channels had no effect on the resting membrane potential even though suprathreshold properties of membrane excitability were significantly changed [19]. This phenomenon is consistent with previous results in hippocampal neurons pretreated with high Ca2+, indicating that membrane potentials may also be targeted by a regulatory mechanism to prevent excitotoxicity [14]. In this study, the enhancement of IDR strongly suggested a neuroprotective mechanism related with IDR channels and Ca2+ influx through VDCCs (Fig. 1). IDR channels contribute to the restoration of depolarized membrane potential to resting membrane potential in cardiac cells, determining refractory period as well as heart rate. In nervous system, these channels seem to contribute to the regulation of somatodendritic excitability. Kv2 family mediated with IDR acts as a dominant regulatory factor for membrane excitability in hippocampal and cortical pyramidal neurons [202122]. Experimental evidence indicates that the genetic manipulation of Kv2.1 directly affects intrinsic excitability, showing epileptiform activity, and that Kv2.1 knock-out mice exhibit defects in spatial learning [23]. These pathogenic outcomes are likely due to the hyperexcitability of neurons during development, as IDR channels are targeted by cellular signaling pathways of various growth factors in young brain [24]. Therefore, enhanced IDR under high Ca2+ condition observed in this study may be a rapid but effective neuroprotective modulation to prevent entry into pathogenic stages.
As a critical regulator of neuronal excitability, IDR channels are dominantly targeted by Ca2+-mediated signaling in the manner of activity- and use-dependence. Even though IDR channels are highly phosphorylated in neurons, the expression and gating kinetics of these channels are bidirectionally subjected to activity-dependent dephosphorylation, which is mediated by glutamatergic transmission [111325]. The dephosphorylation of these channels can rapidly and homeostatically suppress neuronal excitability under pathological conditions via the enhancement of K+ outflow. Dephosphorylation of IDR channels under anoxic stress is dependent on Ca2+ influx through NMDARs that are activated by excess glutamate release in synapses [25]. Thus, overexcitability of glutamatergic synapses in various pathological conditions possibly activates neuroprotective signalings to dephosphorylate and upregulate IDR channels. However, we observed that enhanced IDR under high Ca2+ condition was clearly dependent on Ca2+ influx through VDCCs but not NMDARs. This is evidenced by the effect of nimodipine to abolish the enhancement of IDR (Fig. 1). Despite previous reports on the effects of VDCC blockers to inhibit IDR, nimodipine, one of the dihydropyridines, showed no direct inhibition of IDR in this study (data not shown) [262728]. Because IA downregulation is dependent on NMDARs under high Ca2+ condition, IDR enhancement may be a possible mechanism of compensatory regulation in hippocampal neurons that are chronically exposed to high Ca2+ [14]. IDR channels are predominantly regulated by calcineurin-dependent dephosphorylation, which is accompanied by changes of threshold as well as activation kinetics [18]. In this study, however, we confirmed that high Ca2+-induced upregulation of IDR involved only the increase of IDR peak but not the alteration of gating kinetics, suggesting the possibility of another signaling mechanism distinctive to calcineurin-dependent dephosphorylation. Moreover, the bidirectional and homeostatic upregulation of IDR observed in neurons is due to the overexcitation of glutamatergic synapses, hence, the enhancement of IDR peak without a kinetic change may be originated from Ca2+ influx via VDCCs rather than NMDARs [111325].
The upregulation of IDR peak without changes of gating kinetics suggests the involvement of specific modulatory signalings of kinases, which can be activated under high Ca2+ condition (Fig. 1 and 2). Previous reports focusing on regulatory mechanisms of KV2.1 channel indicate that these channels possess a large number of kinase binding sites such as serine, threonine, and tyrosine, and are usually affected by all except threonine [910]. In particular, tyrosine kinases, such as Src and Fyn, seem to play an important role in the regulation of KV2.1 channel expression. Although, tyrosine 124 phosphorylated by src regulates channels activities, tyrosine 810 phosphorylation influences the intracellular trafficking, regulating Kv2.1 channels expression [293031]. Furthermore, growth differentiation factor 15 (GDF15)-induced upregulation of Kv2.1 channels expression was also abolished by the inhibition of Src-mediated phosphorylation of TGFβ receptors [24]. These previous reports indicate that SFKs are dominantly mediated with the cellular regulation of Kv2.1 channel expression, while the gating kinetics of IDR channels is specifically targeted by serine kinases. Thus, the upregulation of IDR by high Ca2+ treatment observed in this study may be originated from the modulatory function of tyrosine kinases. Fig. 2 clearly shows that PP2 blocked the enhancement of IDR under high Ca2+ condition, indicative of SFKs' roles in IDR channels for preventing hyperexcitability.
Because excessive Ca2+ influx is sufficient to cause hyper-excitability and irreversible damage to hippocampal neurons, resulting in pathological conditions, a number of subcellular signalings in neurons target the expression and gating kinetics of Kv channels which actively regulate membrane excitability. The current study confirmed that a neuroprotective regulatory mechanism involving IDR channels in hippocampal neurons possibly plays a role to suppress neuronal excitability under various pathogenic conditions. Although further study is needed to clarify the relationship(s) between Ca2+ influx via VDCCs and SFK signaling in IDR upregulation, VDCC-mediated upregulation of IDR suggests another signaling pathway to actively regulate membrane excitability of hippocampal neurons.

 XML Download
XML Download