

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Reactive oxygen species (ROS) are formed as a result of partial reduction of oxygen during aerobic respiration [1]. They cause oxidative damage to various biological molecules including DNA, lipids, and proteins, thereby disrupting normal cellular function [234]. Under physiological conditions, ROS are controlled by intracellular free radical scavengers and antioxidant enzymes to protect cells from injuries [5]. However, imbalance between ROS generating and scavenging systems can lead to oxidative stress which can morphologically and functionally damage cells [6]. It is well-known that hydrogen peroxide (H2O2), one type of ROS, can disrupt normal functions in various cell types [23]. It is correlated with overloaded intracellular Ca2+ [789]. However, the mechanism of H2O2-induced Ca2+ accumulation has known to be complicated due to cell-to-cell difference in expression and participation of Ca2+ modulating transporters. It has been reported that H2O2 can enhance Ca2+ release from intracellular store [101112], stimulate Ca2+ entry from extracellular medium [13141516], and attenuate Ca2+ extrusion by plasma membrane Ca2+ ATPase (PMCA) or sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) inactivation [1718] in various cell types.
Pancreatic acinar cells synthesize and secrete a variety of digestive enzyme, tightly regulated by intracellular repetitive Ca2+ oscillation [1920]. A physiological concentration of carbamylcholine (CCh) could generate Ca2+ oscillation known to be initiated by inositol 1,4,5-trisphospate receptors-mediated Ca2+ release from the intracellular store followed by activation of Ca2+ entry from extracellular medium [2122]. The loaded Ca2+ is rapidly cleared to the internal store through SERCA or to the extracellular space through PMCA [23]. Overloaded Ca2+ can cause premature intracellular digestive enzyme activation and cellular injury, one of characteristics of pancreatitis [2425].
Although the pathophysiology of pancreatitis remains unclear at the present time, it has been proposed that oxidative stress due to excess generation of ROS is involved in acute pancreatitis [26]. A prominent feature of acute pancreatitis is disruption of Ca2+ homeostasis within pancreatic acinar cells, and cytosolic Ca2+ accumulation has been shown to cause elevation of ROS in acinar cells that promote cell death [27]. Moreover, there are evidences showing that antioxidants can provide benefits to pancreatitis patients with pancreatic cell injury [28]. However, how ROS accumulates intracellular Ca2+ in pancreatic acinar cell is unclear at the present time. The objective of this study was to characterize the effect of H2O2 on CCh-induced intracellular Ca2+ signals and the underlying mechanism involved in Ca2+ accumulation in mouse pancreatic acinar cells. Here we report that H2O2 could accumulate intracellular Ca2+ by reducing refilling of intracellular Ca2+ stores, rather than by inhibiting Ca2+ extrusion to extracellular fluid or enhancing Ca2+ mobilization from extracellular medium in mouse pancreatic acinar cells.
Go to : 
METHODS
Animals
Male BALB/c mice at 8~10 weeks old were humanely handled and housed under specific pathogen-free conditions in clean polypropylene cages. They were maintained in air conditioned room at 20~22℃ with a constant photoperiod of 12 hours light/dark cycle. Mice were provided free access to pallet diet and drinking water ad libitum. All animal experiments were performed in accordance with the Guideline for the Care and Use of Laboratory Animal provided by NIH. All experiments adhered to Konyang University policies regarding the care and use of animals.
Materials
Type II collagenase was purchased from Roche Diagnostics GmbH (Mannheim, Germany). Fura-2/acetoxymethyl ester (fura-2/AM) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Thapsigargin (TG) was purchased from Tocris (Avonmouth, BS, UK). All other materials were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of pancreatic acinar cells
Small clusters of pancreatic acinar cells (10~15 cells per experiment) were freshly isolated using collagenase digestion method as described previously [2930]. Briefly, the pancreas was removed from mice after CO2 asphyxiation and cervical dislocation. The dissected tissue was enzymatically digested with type II collagenase in HEPES-buffered physiological saline containing 0.01% trypsin inhibitor (soybean) and 0.1% bovine serum albumin (BSA) for 30 minutes followed by mechanical dissociation of cells by gentle agitation. Cells were then filtered through 100 mm nylon mesh and centrifuged at 75 g with 1% BSA. After isolation, cells were resuspended in HEPES-buffered physiological saline containing 137 mM NaCl, 4.7 mM KCl, 0.56 mM MgCl2, 1 mM Na2HPO4, 10 mM HEPES, 1.28 mM CaCl2 and 5.5 mM glucose (pH 7.4 adjusted with NaOH) until use. For Ca2+-free condition, HEPES-buffered physiological saline without adding Ca2+ was supplemented with 5 mM ethylene glycol-bis (2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA).
Intracellular Ca2+ measurements
To measure intracellular Ca2+, the isolated acinar cells were loaded with 5 µM Fura-2/AM and incubated at room temperature in dark condition for 40 minutes. Fura-2/AM loaded cells were mounted onto a cover-glass at the bottom of perfusion chambers. Cells were continuously perfused with HEPES-buffered physiological saline using an electronically controlled perfusion system (Warner Instrument, Hamden, CT, USA). Cells were excited alternately with light at 340 nm and 380 nm using a Polychrome V monochrometer (TILL Photonics, Pleasanton, CA, USA). Fluorescence emission at 505 nm was detected with a Cool-SNAP HQ2 camera (Photometrics, Tucson, AZ, USA) attached to an inverted microscope. The fluorescence ratio of 340/380 was measured using Till-Photonics imaging system. Stimuli were dissolved in HEPES-buffered physiological saline and used to continuously perfuse cells in the perfusion chamber at a flow rate of 1 ml/minute using an electronic controlled perfusion system (Waner Instrument, Hamden, CT, USA).
Data analysis
Values are expressed as mean±SEM. Student t test were used for data analysis. Differences were considered as statistically significant when the p value was less than 0.05. Ca2+ entry rates and extrusion rates were estimated by fitting the increasing and decreasing fluorescence to a single exponential function using Origin program.
Go to :
RESULTS
Effects of hydrogen peroxide (H2O2) on CCh-induced intracellular Ca2+ oscillation
First, the effects of H2O2 on intracellular Ca2+ oscillation were performed in pancreatic acinar cells. Intracellular Ca2+ oscillation was evoked by 500 nM of CCh perfusion in the presence of extracellular Ca2+ at 1.28 mM in intact cells. As shown in Fig. 1A, CCh at 500 nM generated repetitive and sustained Ca2+ oscillation. After the steady state, perfusion of H2O2 at 300 µM resulted in additional elevation of intracellular Ca2+ levels and termination of Ca2+ oscillation in 97±4% cells (n=7, 98 cells). These effects were irreversible even when H2O2 was washed out. Since, in preliminary study, only small proportion of cells were response to H2O2 at 100 µM (34±3%, n=5, 73 cells), we used H2O2 at a concentration of 300 µM in the following studies. Additionally, pretreatment of antioxidants such as catalase at 30µg/ml or 1,4-dithiothreitol (DTT) at 2 mM with CCh completely prevented the effects of H2O2 (i.e., the additional elevation of intracellular Ca2+ levels and the termination of Ca2+ oscillation) (Fig. 1B, C). These results suggest that H2O2 could accumulate intracellular Ca2+ and disrupt normal oscillatory Ca2+ signals in mouse pancreatic acinar cells.
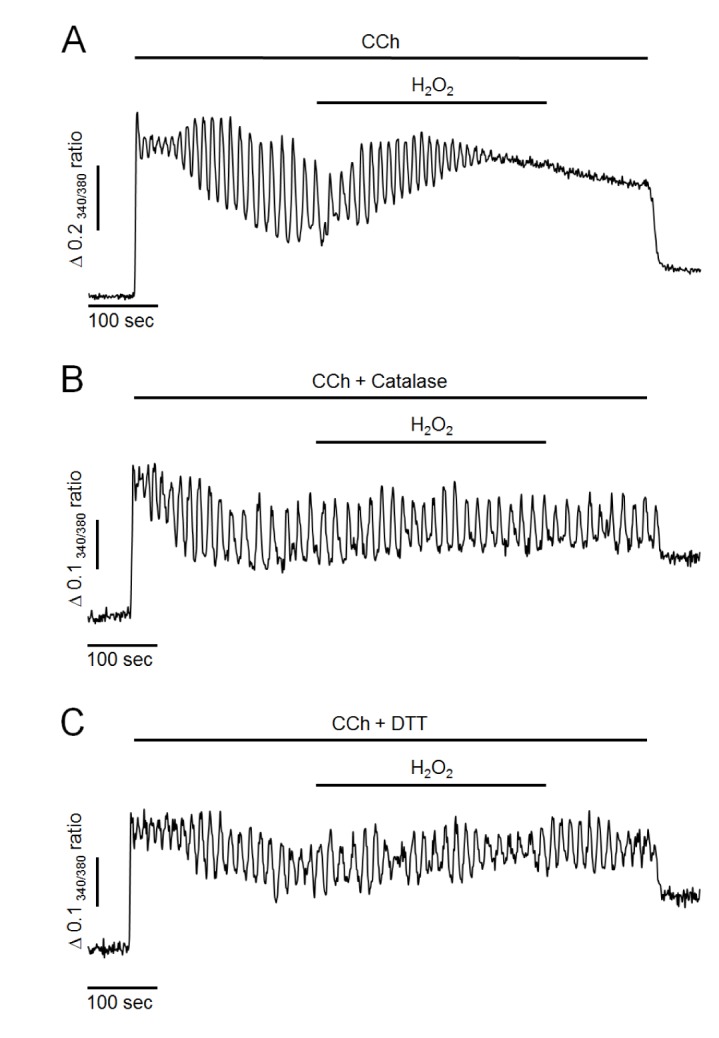 | Fig. 1Effects of hydrogen peroxide (H2O2) and antioxidants on CCh-induced intracellular Ca2+ oscillation in intact pancreatic acinar cells.(A) Representative trace showing the effect of H2O2 on CCh-induced Ca2+ oscillation. (B, C) Representative traces showing the effects of antioxidants (30 µg/ml of catalase and 2 mM of DTT) on H2O2-induced intracellular Ca2+ changes. Oscillatory Ca2+ signals were induced by perfusion with 500 nM of CCh in HEPES buffer containing normal extracellular Ca2+. H2O2 at 300 µM was perfused for 5 minutes. All data were obtained from at least five separate experiments (71~98 cells) and expressed as changes of 340/380 ratio. The perfusion of H2O2 resulted in an elevation of intracellular Ca2+ concentration and a termination of Ca2+ oscillation. Antioxidants completely prevent H2O2-induced Ca2+ accumulation.
|
H2O2 does not affect Ca2+entry or Ca2+extrusion through plasma membrane
Next, we determined whether H2O2-induced Ca2+ accumulation was caused by facilitating Ca2+ entry from extracellular medium or reducing Ca2+ extrusion to extracellular medium through plasma membrane. As shown in Fig. 2A, Ca2+ store was initially depleted with 1 µM of TG in Ca2+-free medium. Store-operated Ca2+ entry was then stimulated by adding extracellular Ca2+ at 1.28 mM. Ca2+ extrusion through plasma membrane was then stimulated by changing to Ca2+-free medium in intact cells. In the control experiment, the adding of extracellular Ca2+ remarkably stimulated Ca2+ entry from extracellular fluid with a Ca2+ entry rate of 0.053±0.009 S−1. H2O2-induced Ca2+ entry rate was 0.056±0.007 S−1, which was not significantly different from the control value (Fig. 2B, C). In H2O2-treated cells, the removing of extracellular Ca2+ clearly extruded intracellular Ca2+ to external space (Fig. 2A). The Ca2+ extrusion rate was 0.033±0.005 S−1, which was not significantly different from its control value at 0.030±0.003 S−1 (Fig. 2D, E). Thus, neither Ca2+ entry from extracellular medium nor Ca2+ extrusion to extracellular medium was modified by H2O2 treatment. Therefore, H2O2-induced Ca2+ accumulation was not due to facilitating Ca2+ entry from extracellular medium or reducing Ca2+ extrusion to extracellular medium through plasma membrane in pancreatic acinar cells.
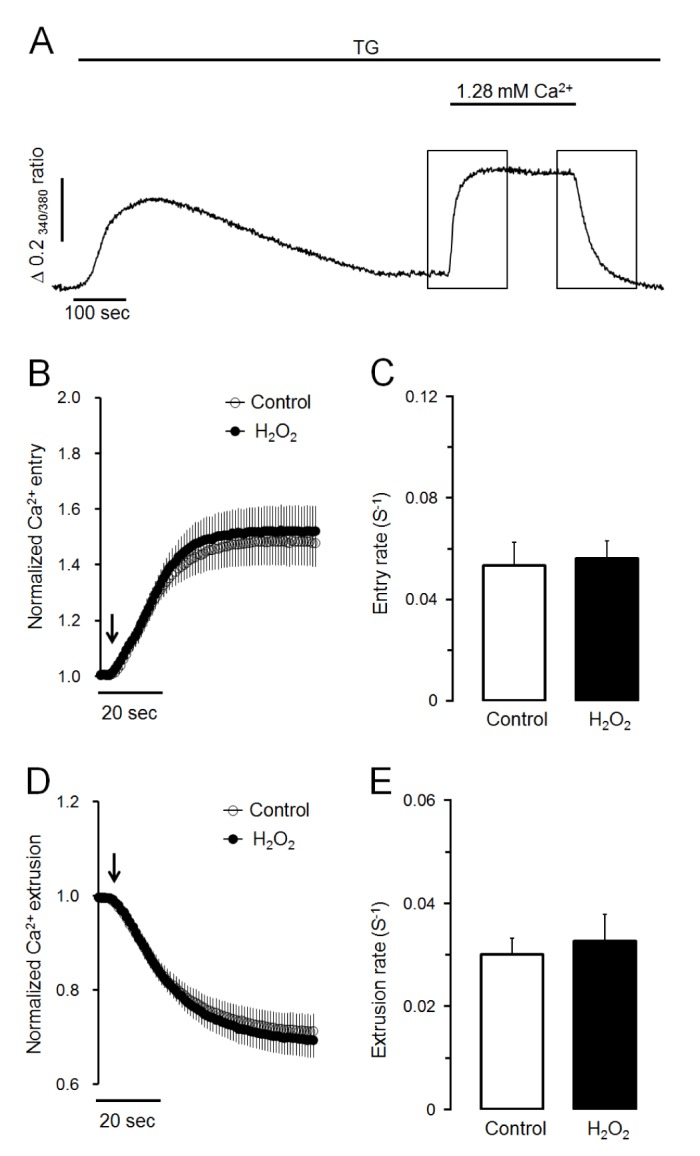 | Fig. 2H2O2 does not affect Ca2+ entry or Ca2+ extrusion in TG-treated pancreatic acinar cells.(A) Representative trace showing the effect of SERCA inactivation using TG on Ca2+ entry from extracellular medium and Ca2+ extrusion to extracellular medium. To deplete intracellular Ca2+ stores, TG at 1 µM was treated in Ca2+-free medium. After depletion of intracellular Ca2+ stores, 1.28 mM of Ca2+ was added and removed to activate Ca2+ entry and Ca2+ extrusion, respectively. (B, C) Effects of H2O2 on normalized Ca2+ entry and Ca2+ entry rate in TGtreated pancreatic acinar cells. Values are expressed as means±SEM obtained from six separate experiments (76 cells). (D, E) Effects of H2O2 on normalized Ca2+ extrusion and Ca2+ extrusion rate in TG-treated cells. H2O2 at 300 µM did not modify Ca2+ entry or Ca2+ extrusion through plasma membrane in TG-treated pancreatic acinar cells.
|
TG mimicsH2O2-induced Ca2+responses and pretreatment of TG completely abolishesH2O2-induced Ca2+responses in Ca2+-free medium
Next, we evaluated whether H2O2 could elevate intracellular Ca2+ levels in Ca2+-free medium because H2O2 did not facilitate Ca2+ entry or reduce Ca2+ extrusion through plasma membrane. As shown in Fig. 3A, in Ca2+-free medium, 500 nM of CCh resulted in Ca2+ oscillation in the initial state, indicating that CCh initially mobilized Ca2+ from intracellular stores. However, oscillatory signals were ceased and returned to baseline levels after 300~500 sec of CCh perfusion in Ca2+-free medium due to discontinued Ca2+ supply from extracellular fluid. After 200 sec of CCh perfusion, treatment with 300 µM H2O2 still resulted in an additional elevation of intracellular Ca2+ levels even when extracellular Ca2+ was eliminated (Fig. 3B). Furthermore, additional elevation of intracellular Ca2+ concentration was mimicked by TG treatment in Ca2+-free medium (Fig. 3C). However, the H2O2-induced additional increase of Ca2+ was completely abolished under SERCA-inactivated condition by TG pretreatment with CCh. Since, in this condition, intracellular TG-sensitive Ca2+ stores were already depleted, TG-insensitive other Ca2+ pools may not participate on H2O2-induced additional elevation of intracellular Ca2+ concentration. These results suggested that H2O2 could accumulate intracellular Ca2+ through inhibiting Ca2+ refilling to intracellular store by inactivation of SERCA, similar to the effect of TG.
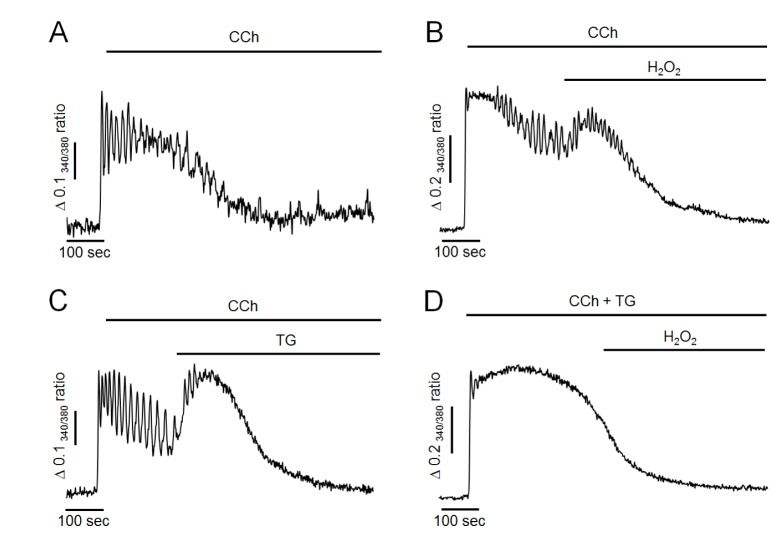 | Fig. 3Effects of H2O2 and TG on CCh-induced intracellular Ca2+ response in Ca2+-free medium.(A) Representative trace showing CCh-induced intracellular Ca2+ response in Ca2+-free medium. (B) H2O2 -induced additional elevation of intracellular Ca2+ levels. (C) TG mimicked the additional elevation of intracellular Ca2+ levels. (D) Pretreatment of TG with CCh completely abolished H2O2-induced additional elevation of intracellular Ca2+ levels. All data were obtained from at least five separate experiments (74~103 cells). Perfusion of H2O2 at 300 µM resulted in additional elevation of intracellular Ca2+ levels, which was mimicked by 1 µM of TG perfusion in Ca2+-free medium. H2O2-induced additional increase of Ca2+ was completely abolished by inactivation of SERCA with TG pretreatment.
|
Ruthenium red does not attenuate H2O2-induced Ca2+responses in Ca2+-free medium
To further determine whether H2O2 could reduce mitochondrial Ca2+ buffering effect, ruthenium red at 50 µM, a mitochondrial Ca2+ uniporter blocker, was used in the following experiment. As shown in Fig. 4A, ruthenium red has no any effect to compare the control experiment (Fig. 3A), and failed to mimic H2O2-induced additional elevation of intracellular Ca2+ levels in Ca2+-free medium. Thus, it is unlikely that mitochondria remarkable participate on Ca2+ elimination after CCh stimulation. Moreover, H2O2 still elevated intracellular Ca2+ levels even when mitochondrial uniporter was blocked by pretreatment of ruthenium red with CCh (Fig. 4B). This result indicated that Ca2+ accumulation induced by H2O2 might not be by reducing Ca2+-buffering capacity of mitochondria in pancreatic acinar cells.
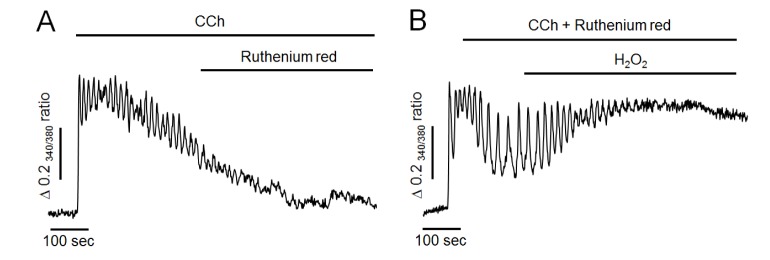 | Fig. 4Effect of ruthenium red on H2O2-induced intracellular Ca2+ response in Ca2+-free medium.(A) Representative trace showing the effect of ruthenium red on CCh-induced intracellular Ca2+ response in Ca2+-free medium. (B) Pretreatment of ruthenium red with CCh failed to attenuate H2O2-induced additional elevation of intracellular Ca2+ levels. All data were obtained from six and seven separate experiments (70 and 81 cells). Perfusion of ruthenium red at 50 µM, a mitochondrial Ca2+ uniporter inhibitor, did not mimic H2O2-induced additional elevation of intracellular Ca2+ levels. After pretreatment of ruthenium red with CCh, H2O2 still elevated intracellular Ca2+ concentration.
|
Go to :
DISCUSSION
The present study clearly provided evidence that H2O2, a reactive oxygen species, could accumulate cytosolic Ca2+ through attenuating refilling of intracellular Ca2+ store in mouse pancreatic acinar cells. Cytosolic free Ca2+ plays a pivotal role in the stimulus-secretion coupling process in pancreatic acinar cells [1920]. Ca2+ can be mobilized to elicit physiological responses from both the external fluid and the internal stores such as endoplasmic reticulum and acidic store. Acetylcholine and cholecystokinin (CCK), the major agonists in pancreatic exocrine gland, are known to generate repetitive and transient oscillatory Ca2+ signals [2122]. The balance between Ca2+ mobilization and Ca2+ elimination is important to generate Ca2+ oscillation in exocrine cells. These processes are regulated by the action of a variety of channels, pumps, and exchangers for Ca2+ localized both in the plasma membrane and the ER membrane [1920]. Since the accumulation of intracellular Ca2+ causes cellular damage associated with acute and chronic pancreatitis [2425], basal intracellular Ca2+ concentrations have to be finely regulated to low resting values under normal condition.
Although oxidant-induced intracellular Ca2+ overload has been revealed in various cell types, the underlying mechanisms of Ca2+ mobilization and elimination are complicated [1011,12,13,14,15,16,1718]. It has been known that the involvement of oxidants in Ca2+ homeostasis is mediated by the modification of disulfide bonds between cysteine residues of Ca2+ regulating proteins including SERCA, PMCA and Na+/Ca2+ exchanger (NCX) [3132]. These molecules have different isoforms with different expression characteristics and regulation properties, thus giving versatility of Ca2+ signaling [33]. The present study was designed to investigate the exact mechanism of how H2O2 could cause intracellular Ca2+ accumulation in pancreatic acinar cells. When acinar cells were exposed to 300 µM of H2O2 in normal buffer, there was additional elevation of cytosolic Ca2+ and termination of oscillatory Ca2+ signals. These effects of H2O2 on Ca2+ signals were completely prevented by pretreatment with catalase (an enzyme that can degrade hydrogen peroxide) and DTT (a sulfhydryl reducing agent). Although cytosolic H2O2 concentrations produced by oxidative stress in pancreatic acinar cells is not known, only small proportion of cells (34%) were response to 100 µM of H2O2 and most cells (97%) were response to 300 µM of H2O2 in the present study. In general, H2O2 at concentrations from 10 µM to 1 mM caused intracellular Ca2+ accumulation in various cell types [1011,12,13,14,15,16,17]. These results suggest that excess generation of oxidants in pathologic conditions could disturb Ca2+ homeostasis mediated by sulfhydryl group oxidation in pancreatic acinar cells.
Next, we investigated whether H2O2 actually induced Ca2+ entry from extracellular fluid through plasma membrane. In ventricular myocyte, ROS can enhance Ca2+ entry through modulating the function of voltage-gated L-type Ca2+ channels in plasma membrane [34]. It has been reported that ROS play physiological roles in platelet aggregation by activating SOC-mediated Ca2+ entry in human platelets [35]. Transient receptor potential (TRP) channels, such as TRPC3, TRPM2, TRPM7, and TRPPA1 are also known to sensitive to ROS [36]. They participate in neurodegeneration process of neuronal cells. However, H2O2-induced Ca2+ accumulation still occurred in Ca2+-free medium in this study after extracellular Ca2+ sources were eliminated. Furthermore, H2O2 failed to attenuate SOC-mediated Ca2+ entry by adding extracellular Ca2+ at 1.28 mM after ER Ca2+ stores were depleted in Ca2+-free medium by pretreatment of TG. In pancreatic acinar cells, evidence of the existence or the role of voltage-gated Ca2+ channels or NCX has not been presented. The role of TRP channels and their sensitivities to H2O2 have also not been fully elucidated at the present time. Our results suggested that Ca2+ entry channels in plasma membrane might not be the primary targets of H2O2-induced Ca2+ accumulation in mouse pancreatic acinar cells.
In this study, H2O2-induced additional elevation of intracellular Ca2+ concentration was mimicked by TG treatment in Ca2+-free medium. Moreover, H2O2-induced additional increase of Ca2+ was completely abolished in SERCA-inactivated condition by TG pretreatment with CCh. These results strongly suggest that H2O2 could accumulate intracellular Ca2+ through inhibiting refilling to intracellular Ca2+ store, similar to the effect of TG by inactivating SERCA. Since SERCA contains 20~28 cysteine residues, its activity can be effectively modulated by oxidants. It has been reported that ROS could attenuate the activity of this pump by modifying sulfhydryl groups [37]. Distinct SERCA isoforms are known to show different susceptibilities to ROS due to different location of cysteine residues [38]. In rat pancreatic acinar cell, there was no expression of SERCA1 mRNA and SERCA2 mRNA expression was down-regulated in acute pancreatitis [39]. The different sensitivity to H2O2 between SERCA subtypes is not known at the present time. Thus further studies are needed to elucidate the mechanism of H2O2 on calcium accumulation and cell damage. PMCA also could contribute to ROS-induced cytosolic Ca2+ accumulation because this pump has abundant cysteine residues [37]. In this study, 300 µM of H2O2 failed to attenuate Ca2+ extrusion through plasma membrane in TG-treated experiment. However 10 folds higher concentration of H2O2 partially inhibited Ca2+ extrusion to extracellular fluid under similar conditions (data not shown). These findings strongly suggest that the primary target for H2O2-induced Ca2+ accumulation might be SERCA rather than PMCA in mouse pancreatic acinar cells.
The overloaded Ca2+ also could be eliminated by buffering action of mitochondria. CCK can evoke oscillatory Ca2+ signals and substantial mitochondrial Ca2+ uptake in pancreatic acinar cells [4041]. H2O2 can cause mitochondrial Ca2+ release abolished by pretreatment of FCCP or CCCP, a mitochondrial uncoupler [4041]. However, in another study, mitochondrial Ca2+ uptake did not occur in unstimulated resting cells [18]. In addition, H2O2-induced mitochondrial Ca2+ uptake was very slow at low capacity even cells were stimulated by CCK [18]. In the present study, ruthenium red alone has no effect on CCh-induced Ca2+ response in Ca2+ free medium, and H2O2 still elevated intracellular Ca2+ levels even when mitochondrial uniporter was blocked by pretreatment of ruthenium red with CCh. Thus, it is unlikely that mitochondria are the major source of H2O2-induced elevation of cytosolic Ca2+.
Based on the above results, we conclude that the primary target molecule for excessively generated H2O2 in pathological conditions is likely to be the sulfhydryl group of SERCA. We also conclude that H2O2 can accumulate intracellular Ca2+ by attenuating the refilling of intracellular Ca2+ stores through ER membrane rather than by Ca2+ entry or Ca2+ extrusion through plasma membrane in mouse pancreatic acinar cells.
Go to :
 XML Download
XML Download