

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Tricyclic antidepressants (TCAs), including amitriptyline, butriptyline, nortriptyline, clomipramine, desipramine, and doxepin, have been the first selection for pharmacological treatment of clinical depression for many years [12]. Nortriptyline is second-generation TCA and is mainly used in the treatment of major depression and childhood nocturnal enuresis [3]. Additionally, although nortriptyline has been shown to have fewer side effects than tertiary amine TCAs, such as amitriptyline, imipramine, and clomipramine, some side effects, including dry mouth, sedation, constipation, mild blurred vision, tinnitus, euphoria, and mania, have still been observed [4]. However, to date, the side effects of nortriptyline on vascular smooth muscle, specifically ion channels, are unknown.
The alteration of membrane potential is regarded as a major determinant of changes in vascular diameter, and can thereby alter blood pressure and organ blood flow [56]. Several ion channels expressed in arterial smooth muscle, including Ca2+, K+, and Cl− channels, are involved in maintaining and changing the membrane potential. Of these, K+ channels play the most crucial role in determining the resting membrane potential of arterial muscle cells. In arterial muscle cells, four types of major K+ channels have been detected: ATP-sensitive K+ (KATP), large-conductance Ca2+-activated K+ (BKCa), inwardly rectifying K+ (Kir), and voltage-dependent K+ (Kv) channels [57]. Although most K+ channels play essential roles in maintaining resting membrane potential, Kv channels in particular are regarded as the most crucial channels for regulating resting membrane potential [8]. In fact, inhibition of Kv channels in some arteries induces strong membrane depolarization [910]. Additionally, reduction of Kv channel activity and/or expression has been identified in various metabolic and circulatory diseases [8111213]. Furthermore, the modulation of vascular Kv channel is closely related to various intracellular protein kinases, such as protein kinase C (PKC), protein kinase A (PKA), and protein kinase G (PKG) [7]. Therefore, the unexpected effects of some agents on vascular Kv channels must be clearly identified to reduce potentially toxic vascular effects.
In this study, we demonstrated the inhibitory effect of nortriptyline on Kv channels using native coronary arterial smooth muscle from rabbits. We found that nortriptyline inhibits Kv channels in a concentration-dependent and state-independent fashion independently of serotonin reuptake inhibition.
Go to : 
METHODS
Single cell isolation
In accordance with the guidelines of the Committee for Animal Experiments of Kangwon National University, male New Zealand White rabbits (2.0~2.5 kg) were anesthetized using pentobarbitone sodium (40 mg/kg) and heparin (120 U/kg) injected simultaneously into the ear vein. The chest was opened, the heart separated and left descending coronary arteries were dissected and collected in normal Tyrode's solution. To obtain a suspension of single smooth muscle cells, the arteries were digested in 1 ml Ca2+-free normal Tyrode's solution containing papain (1.0 mg/mL), bovine serum albumin (BSA, 1.0 mg/mL), and dithiothreitol (DTT, 1.0 mg/mL) for 24 min at 37℃. After this incubation step, the solution was replaced with 1 ml Ca2+-free normal Tyrode solution containing collagenase (2.8 mg/mL), BSA, and DTT, for 22 minutes at 37℃. A suspension of single smooth muscle cells was obtained by gentle agitation of the digested arteries using a pasture pipette and kept at 4℃ in Kraft-Brühe (KB) solution. Isolated cells were preserved at 4℃ for use in experiments within 6 h.
Solutions and chemicals
The composition (in mM) of normal Tyrode's solution was: CaCl2, 1.8; NaCl, 135; NaH2PO4, 0.33; KCl, 5.4; HEPES, 5; MgCl2, 0.5; glucose, 16.6; adjusted to pH 7.4 with NaOH. The composition (in mM) of KB solution was: KCl, 55; KOH, 50; KH2PO4,70; taurine, 20; L-glutamate, 20; MgCl2, 3; HEPES, 10; glucose, 18; EGTA, 0.6; adjusted to pH 7.3 with KOH. The composition (in mM) of the pipette solution was: KCl, 25; K-aspartate, 110; NaCl, 5; Mg-ATP, 4; MgCl2, 2; HEPES, 10; EGTA, 10; adjusted to pH 7.2 with KOH. Nortriptyline was purchased from Sigma Chemical Co. (St. Louis, MO, USA) and dissolved in distilled water. DPO-1 and guangxitoxin were purchased from Tocris Cookson (Ellisville, MO, USA) and dissolved in dimethyl sulfoxide (DMSO).
Electrophysiological recordings and data analysis
Kv current were recorded in individual vascular smooth muscle cells using PatchPro software, NI-DAQ-7 digital interface (National Instruments, Union, CA, USA), and EPC-8 amplifier (Medical system Corp., Darmstadt, Germany). Borosilicate capillary glass pipettes (Clark Electromedical Instruments, Pangbourne, UK) were pulled on a PP-830 puller (Narishige Scientific Instrument Laboratory, Tokyo, Japan) with a resulting resistance of 3~4 MΩ.
Data analysis was completed using Origin 7.0 software (Microcal Software, Inc., Northampton, MA, USA). The drug-channel interaction kinetics is described as a first-order blocking model [14]. The values for the half-maximal inhibitory (IC50) and the slope value (n) were calculated from concentration-dependent data and fitted to the Hill equation:
where ƒ represents the fractional current inhibition (ƒ= 1-Idrug/Icontrol) at each potential, and [D] represents drug concentration.
Channel activation was calculated from depolarizing from −80 to +60 mV in 10-mV increment returning to −40 mV. The recorded tail currents were normalized to the maximal tail current at each depolarizing step and fitted with the Boltzmann equation:
where V is the test potential, V1/2 represents the half-point of activation, and k is the slope factor.
Steady-state inactivation was calculated using a two-step voltage protocol from a test potential of +40 mV for 600 ms after 7-s of preconditioning pulses applied at potentials ranging from −80 to +30 mV in the absence and presence of nortriptyline. The steady-state inactivation curve was calculated from another Boltzmann equation:
where V is the potential for preconditioning pulses, V1/2 is the potential of the mid-maximal of inactivation, and k represents the slope value.
All data is expressed as means±standard error of the mean (S.E.M) and the Student's t-test was applied to determine significance with p<0.05 regarded as statistically significant.
Go to :
RESULTS
Inhibition of Kv current by nortriptyline
The inhibitory effects of nortriptyline were recorded using native coronary smooth muscle cells from rabbit heart. The involvement of other vascular K+ channels was prevented by inclusion of EGTA (used for blocking BKCa channels) and ATP (used for blocking KATP channels) in the internal solution. In addition, activation of Kir channels was excluded by using large-diameter arterial smooth muscle cells, as the Kir channel has only been identified in small-diameter arteries [15]. Kv currents were induced by 10-mV step-depolarizing pulses from −60 to+60 mV from −60 mV holding potential. As shown in Fig. 1A, the control Kv current rapidly ascended to a peak and slowly and partially inactivated due to intrinsic inactivation. Application of 10 µM nortriptyline reduced the elicited Kv current within 1 min (Fig. 1B). This inhibition was partially washed out (~60%). Fig. 1C shows the current-voltage (I~V) relationship of the steady-state current in the presence or absence of 10 µM nortriptyline. At +60 mV, nortriptyline reduced the Kv current by 43.35%. To test the voltage-dependency of Kv channel inhibition by nortriptyline, current (Idrug/Icontrol) was plotted as a function of membrane potential using I~V relationship curve. As shown in Fig. 1D, nortriptyline inhibited Kv currents in a voltage-independent manner.
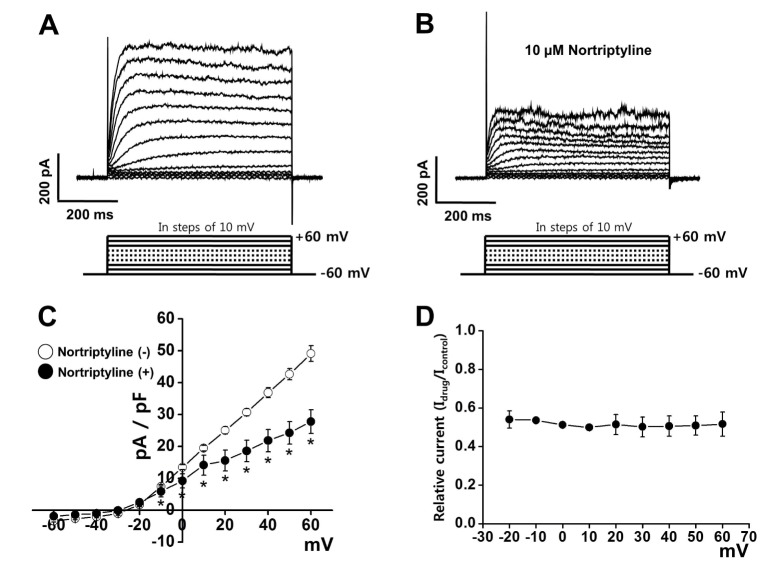 | Fig. 1Effects of nortriptyline on voltage-dependent K+ (Kv) channel currents.Representative Kv currents were elicited by 600-ms depolarizing step pulses from −60 to +60 mV in steps of 10-mV at a holding potential of −60 mV in the control condition (A) and in the presence of 10 µM nortriptyline (B). (C) The current-voltage (I~V) relationship at steady-state Kv current under control conditions (○) and in the presence of 10 µM nortriptyline (●). n=7. *p<0.05. (D) Voltage-dependence of Kv current inhibition by nortriptyline. Normalized inhibition shown as relative current from panel in (C) are plotted as a function of membrane potential.
|
Nortriptyline inhibits the Kv current in a dose-dependent manner
Several concentrations (0, 0.1, 0.3, 1, 3, 10, 30, and 100 µM) of nortriptyline were applied to determine whether the nortriptyline-induced inhibition of Kv channels was dose-dependent. The superimposed currents were obtained by applying +60 mV repetitive depolarizing pulses from −60 mV holding potential. As shown in Fig. 2B, Kv channel inhibition increased with increasing concentrations of nortriptyline. The recorded currents at each concentration were normalized to currents recorded in the absence of nortriptyline, and were fitted to the Hill equation (Fig. 2B). From this, we obtained an IC50 value of 2.86±0.52 µM and a slope value of 0.77±0.10. These results suggest that nortriptyline inhibits Kv channels in a dose-dependent manner.
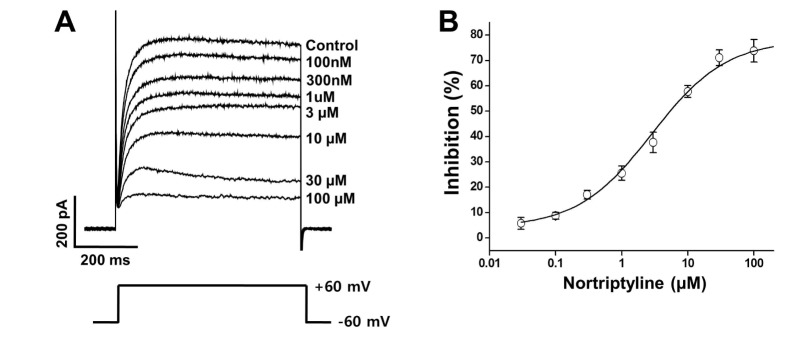 | Fig. 2Concentration-dependent inhibition of Kv channels by nortriptyline.(A) Superimposed currents were elicited by 600-ms depolarizing pulse of +60 from a holding potential of −60 mV in the presence of 0, 0.1, 0.3, 1, 3, 10, 30, and 100 mM nortriptyline. (B) Concentration-dependent curve for nortriptyline-induced inhibition of the Kv current measured at steady-state. n=8.
|
Effects of nortriptyline on steady-state activation and steady-state inactivation of the Kv current
To determine whether nortriptyline affects the gating properties of Kv channels by changing the steady-state activation and/or inactivation curves, we investigated the nortriptyline effect on the activation and inactivation kinetics of Kv channels. Steady-state activation curves were calculated using relative tail currents, and the results were fit to a Boltzmann equation. As shown in Fig. 3A, applying 10 µM nortriptyline did not affect the activation curve. The mid-maximal potential (V1/2) and slope values (k) were 4.98±0.76 mV and 4.82±0.69 under control conditions and 4.35±0.68 mV and 4.52±0.43 in the presence of nortriptyline, respectively.
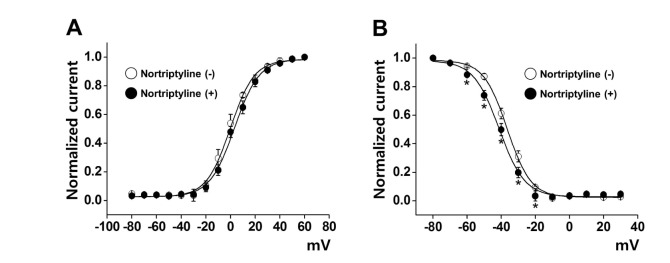 | Fig. 3Influence of nortriptyline on steady-state activation and steady-state inactivation curves.(A) Activation currents in the absence (○) and presence of 10 µM nortriptyline (●). Tail currents were elicited by short depolarizing pulses (20~50 ms) from −80 to +60 mV in steps of 10 mV at a holding potential of −80 mV. Subsequent pulses returned to −40 mV. The current was normalized from the peak value of the tail current. n=10. (B) Inactivation currents in the absence (○) and presence of nortriptyline (●). The currents were elicited by a two-pulse protocol with a test step to +40 mV after 7-s conditioning pre-pulses between −80 and −30 mV. The elicited currents were normalized to the current of the peak test pulse. n=9. *p<0.05.
|
We also investigated the influence of nortriptyline on steady-state inactivation curves. The pulse protocol to yield steady-state inactivation curves is described in the Materials and Methods. The steady-state inactivation curves were fit to another Boltzmann equation. As shown in Fig. 3B, application of 10 µM nortriptyline moved the inactivation curve toward a more negative potential. The mid-maximal potential (V1/2) and slope value (k) were −39.22±2.29 mV and 5.89±0.96 under control conditions and −43.68±1.37 mV and 6.88±0.84 in the presence of 10 µM nortriptyline, respectively.
Effects of nortriptyline on Kv channels in the presence of Kv1.5 and/or Kv2.1/2.2 inhibitors
To determine the contribution of the Kv1.5 and/or Kv2.1/2.2 currents to nortriptyline inhibition, we tested the nortriptyline effect on Kv channels in the presence of a Kv1.5 inhibitor (DPO-1, 1 µM) and/or a Kv2.1/2.2 inhibitor (guangxitoxin, 30 nM) by application of a +60 mV depolarizing pulse from −60 mV holding potential. As shown in Fig. 4A~D, addition of DPO-1 or guangxitoxin quickly decreased the Kv current. Application of nortriptyline additionally induced further Kv current inhibition. However, the degree of further inhibition was similar to nortriptyline alone. In addition, simultaneous pre-treatment with DPO-1 and guangxitoxin had no significant effect on the degree of Kv channel inhibition by nortriptyline (Fig. 4E and F). From these results, we speculate that Kv1.5 and Kv2.1/2.2 are not the main targets of nortriptyline.
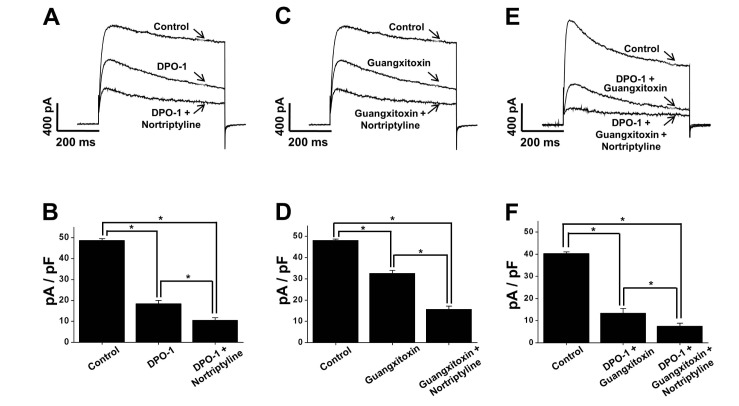 | Fig. 4The effects of pre-treatment with Kv1.5 and/or Kv2.1/2.2 inhibitors on the nortriptyline-induced inhibition of Kv current.Current traces were obtained by one-step depolarizing pulses to +60 mV at a holding potential of −60 mV. (A) Superimposed currents under control, in the presence of DPO-1, and in the presence of DPO-1+nortriptyline. (B) Summary of the results shown in panel (A). n=5. *p<0.05. (C) Superimposed currents under control, in the presence of guangxitoxin, and in the presence of guangxitoxin+nortriptyline. (D) Summary of the results shown in panel (C). n=5. *p<0.05. (E) Superimposed currents under control, in the presence of DPO-1+guangxitoxin, and in the presence of DPO-1+guangxitoxin+nortriptyline. (F) Summary of the results shown in panel (E). n=6. *p<0.05.
|
Use-dependency of Kv current by nortriptyline
To evaluate the use-dependent inhibition of Kv channels by nortriptyline, we applied 20 repeated one-step depolarizing pulses to +60 mV at frequencies of 1 or 2 Hz. The train repetitive pulses are frequently used to examine the use- and/or state-dependent inhibition of ion channels. As shown in Figs. 5A and B, the Kv current decreased progressively during the repetitive depolarizing pulses. However, this progressive decrease in Kv current was not changed by application of 10 µM nortriptyline. These results suggest that nortriptyline-induced inhibition of Kv channels is not use-dependent.
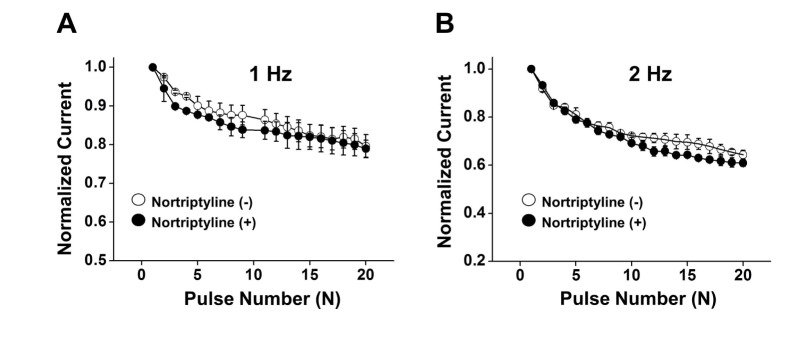 | Fig. 5Frequency-dependent inhibition of Kv currents by nortriptyline.Twenty depolarizing pulses from −60 mV to +60 mV in duration of 150-ms were applied in the absence (○) and presence of nortriptyline (●), at frequencies of 1 Hz (A) and 2 Hz (B). Each peak current was normalized by the peak current at first pulse and plotted against the pulse number. n=5.
|
Go to :
DISCUSSION
In this study, we demonstrated the inhibitory effect of nortriptyline, a member of TCAs, on the Kv channel current in native coronary smooth muscle cells from rabbit. Our study suggested that nortriptyline inhibited vascular Kv channels in a dose-dependent and state-independent manner regardless of serotonin reuptake inhibition. In addition, nortriptyline shifted the steady-state inactivation curves toward a negative potential.
Based on our results, we speculated that nortriptyline inhibited Kv channels independently of serotonin reuptake inhibition. First, the IC50 value of nortriptyline for Kv current inhibition was 2.86±0.52 µM, which was much greater than the IC50 value of nortriptyline for serotonin uptake inhibition (0.94 µM) [16]. This difference suggests that the nortriptyline-induced inhibition of Kv channels is independent of serotonin uptake inhibition. Second, nortriptyline-induced inhibition of Kv channels occurred within 1 min. This fast interaction between nortriptyline and vascular Kv channels suggested that Kv channel inhibition by nortriptyline was not related with complicated intracellular signaling pathways, but rather by direct interaction. Third, as described in Fig. 3, nortriptyline shifted the steady-state inactivation curve of Kv channels leftward; suggesting that nortriptyline directly interacted with Kv channels. Fourth, increasing serotonin levels in the blood stream could increase cardiovascular risk, especially in hypertensive patients [17], since serotonin inhibited Kv channels in arterial smooth muscle [10]. However, nortriptyline-induced increases in serotonin levels were not possible in our single cell experimental system. Therefore, nortriptyline effect on Kv channels was completely independent of nortriptyline-induced increases in serotonin levels. These results suggest that the inhibitory effect of nortriptyline on Kv channels is not related to serotonin reuptake inhibition, but is more likely due to a direct interaction.
Nortriptyline may bind to the Kv channels in a state-independent manner and reduces channel availability or decreases channel conductance. The binding site of Kv channels for nortriptyline could be either near channel pore or at other regulatory non-pore region. It is possible that nortriptyline allosterically binds to non-pore region of Kv channels and induces conformational changes in Kv channels, rendering them less available and/or with less conductance. Provided that this allosteric binding affects the voltage-dependency of Kv inactivation, the latter possibility may provide ratiomale for the leftward shift of steady-state inactivation curve of Kv channels in the presence of nortriptyline.
Nortriptyline is second-generation TCA and is effective for the treatment of major depression and childhood nocturnal enuresis [3]. Although nortriptyline has fewer and milder side effects than tertiary TCAs (e.g., imipramine and amitriptyline) and therefore used more preferentially than other TCAs, the common side effects including sedation, dry mouth, mild blurred vision, constipation, increased appetite, tinnitus, often euphoria and mania have been detected [18]. Several side effects relating to the effect of nortriptyline on ion channels have also been reported. For example, nortriptyline inhibited astroglial inwardly rectifying Kir4.1 channels expressed in human embryonic kidney 293 (HEK293) cells. This inhibition occurred in a dose-, time-, and voltage-dependent manner [19]. Nortriptyline also inhibited human small conductance calcium-activated potassium channels (hSK3) expressed in CHO-K1 cells. This study suggests that TCAs, including nortriptyline, inhibit hSK3 channels by directly interacting with the channel. Although the detailed binding mechanisms remain unknown, the common three-ring core structure of TCAs seems to be crucial for channel inhibition [20]. In addition, nortriptyline reportedly inhibits human ether-a-go-go-related gene (hERG) channels expressed in Xenopus oocytes [21] and cardiac sodium channels [22]. Although several papers have attempted to identify the side effects of nortriptyline on ion channels, the effects of nortriptyline on Kv channels in native vascular smooth muscle cells have been neglected. In this study, we investigated the inhibitory effect of nortriptyline on native vascular smooth muscle cells.
Kv channels are highly expressed in most arterial smooth muscle cells, and they mainly participate in regulating membrane potential and vascular diameter [85]. Alteration of Kv channel activity and/or expression is closely associated with several pathological conditions, including hypertrophy, hypertension, diabetes, and hypoxia [8]. Therefore, it is important to identify the side effects of some drugs on vascular Kv channels to reduce misunderstanding of experimental data. Many drugs inhibit the vascular Kv channel regardless of their own function, and these experiments have mainly been performed in our laboratory. We recently reported that fluvoxamine, a selective serotonin reuptake inhibitor, had inhibitory action on vascular Kv channels. Consistent with this study, fluvoxamine shifted the inactivation curve to a more negative potential without altering activation curve [23]. Furthermore, cisapride, a serotonin 5-HT4-receptor agonist, also inhibited vascular Kv channels by shifting the inactivation curve toward a negative potential [24]. In addition, two selective serotonin reuptake inhibitors, paroxetine and sertraline have been reported to inhibit Kv 1.5 channels in CHO cells [2526]. In this study, we clearly demonstrated the inhibitory effect of nortriptyline, a serotonin reuptake inhibitor, on vascular Kv channels. Considering the clinical efficacy of nortriptyline and pathophysiological relevance of arterial Kv channels, our results should be considered when prescribing nortriptyline in patients with depression.
Although previous studies agree that Kv1.1, Kv1.2, Kv1.4, Kv1.5, Kv2.1, and Kv9.3 subtypes are detected in arterial smooth muscle cells [62728], the Kv channel subtypes expressed in rabbit arterial smooth muscle cells were still unknown. This is due to the fact that most previous studies on Kv subtype expression in arterial smooth muscle have mainly used rat, mouse, and human samples. For this reason, it is difficult to describe exactly which Kv subtypes were involved in the nortriptyline-induced inhibition of Kv channels. However, we tested the involvement of Kv1.5 and Kv2.1/2.2, which are known as the major Kv subtypes in vascular smooth muscle, in nortriptyline-induced inhibition of Kv channels by applying pharmacological inhibitors. As shown in Fig. 4, Kv1.5 and Kv2.1/2.2 inhibitors had no effect on the inhibitory effect of nortriptyline on Kv channels. Although we could not exactly address the specific Kv subtypes involved in nortriptyline-induced inhibition of Kv channel, the inhibitory effect of nortriptyline may not be simply related to the inhibition of a single Kv subtype, but may involve multiple Kv subtypes.
Although DPO-1 is a specific inhibitor of Kv1.5 channels (IC50 value of 0.31 µM) [29], it may lose selectivity over subtypes of Kv channels with extremes of the dose range. For example, DPO-1 is known to inhibit the Kv3.1 channel expressed in Xenopus oocytes when used at 15-fold higher concentrations (4.7 µM) [29]. In addition, DPO-1 inhibited Kv1.3 subtype with IC50 values of 2.58 µM in Jurkat cells and 3.11 µM in human peripheral blood T cells [30]. Therefore, the DPO-1 concentration (1 µM) used this study could affect some portion of Kv3.1 and Kv1.3 channel activity. However, considering that IC50 value for Kv1.5 subtype inhibition was 0.31 µM and most previous studies have applied 1 µM DPO-1 to inhibit Kv1.5 subtype [3132], our experimental concentration of DPO-1 is the best and suitable concentration for the inhibition of Kv1.5 subtype.
Clinically, nortriptyline is prescribed in 25 mg doses 3 or 4 times daily. The plasma concentration of nortriptyline reaches a peak value (31.35 nM) with 7 h of taking a 25 mg tablet [33]. In this study, we clearly identified that the IC50 value for inhibition of Kv channels by nortriptyline was 2.86±0.52 µM, which is greater than the plasma peak concentration of nortriptyline in the blood. However, our results showed that lower concentrations of nortriptyline (100 nM) also effectively inhibited Kv channels. Therefore, an overdose of nortriptyline could increase plasma concentrations to greater than 100 nM. Considering that vascular smooth muscles maintain high input resistance, changes of only a few picoamperes could induce changes in vascular tone, and consequently in blood flow and pressure [34]. Although nortriptyline has therapeutic efficacy for the treatment of major depression, strict regulation of nortriptyline dosing is necessary, especially in patients with cardiovascular diseases.
In summary, this study demonstrated that nortriptyline inhibited the vascular Kv channels in a concentration-dependent and state-independent manner. Considering the clinical efficacy of nortriptyline for the treatment of depression and the physiological importance of Kv channels in vascular function, our findings should be considered when prescribing nortriptyline to patients with depression.
Go to :
 XML Download
XML Download