

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Status epilepticus (SE) is the most common serious neurological condition with respect to morbidity and mortality, with a prevalence of 0.5% [1]. SE is triggered by abnormal electrical activity, leading to delayed cell death in the brain [2]. Although many studies have attempted to develop new antiepileptic drugs that could be effective in terminating SE and preventing its long-term consequences [3], more than a third of patients with epilepsy suffer from uncontrolled seizures that lead to substantially diminished quality of life.
Lithium has been extensively used for decades as a mood stabilizer in manic depressive disorders [4]. In addition to its antimanic effects, lithium has shown anticonvulsant activity in several animal models of seizures [5678]. Lithium has also been identified as a potential neuroprotective agent against neurodegeneration, although the mechanism for this has not been completely clarified. The neuroprotective properties of lithium have been reported in focal cerebral ischemia [9] and several in vitro injury models [10].
The pilocarpine model in mice is considered the most suitable experimental model of temporal lobe epilepsy. The muscarinic receptor agonist pilocarpine is used to induce SE, which is followed by its neuropathological features, such as neuronal death, reactive gliosis, and remodeling of synaptic circuitry. In combination with pilocarpine, lithium pre-treatment potentiates the epileptogenic action of pilocarpine and allows a reduction of the pilocarpine dose required to elicit SE. The syndromes triggered by pilocarpine and lithium-pilocarpine in mice have been shown to be behaviorally and neuropathologically similar [11]. Considering the proconvulsive activity of lithium when acting in combination with pilocarpine, it would be reasonable to investigate how acute administration of lithium after pilocarpine injection could alter sequential behavioral changes and neuronal damage resulting from pilocarpine-induced SE.
In the present study, we investigated the effect of lithium post-treatment on seizure susceptibility and hippocampal damages following pilocarpine-induced SE. Lithium post-treatment following pilocarpine-induced SE delayed the onset time of SE and reduced mortality and neuronal injury.
Go to : 
METHODS
Chemicals
Lithium chloride, distrene plasticizer xylene (DPX), pilocarpine hydrochloride, potassium permanganate and cresyl violet acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA) and atropine methyl nitrate was obtained from Tokyo Chemical Industry Co. (Tokyo, Japan). Fluoro-Jade B and glial fibrillary acidic protein (GFAP) were purchased from Millipore (Temecula, CA, USA). The CD11b antibody came from Abcam (Cambridge, MA, USA).
Pilocarpine-induced status epilepticus model
The pilocarpine model of SE in mice was previously described [1213]. Briefly, male C57BL/6 mice (7~8 weeks of age) were administered atropine methyl nitrate (1.2 mg/kg, i.p.) 30 min before the injection of pilocarpine hydrochloride (320 mg/kg, i.p.). After pilocarpine administration, the behavior of the mice was closely monitored for approximately 6 h to evaluate the onset time of stage 4 seizure, SE, severity, and mortality. SE was defined as a continuous motor seizure at stage 4 (rearing and falling), stage 5 (loss of balance, continuous rearing and falling) and stage 6 (severe tonic clonic seizures) (Racine, 1972) [14]. In this study, only mice that showed severe tonic-clonic seizures were included. After 15 min of pilocarpine administration, lithium chloride (80 mg/kg, i.p.) (n=29) or vehicle (saline, n=31) was administered. The mice were sacrificed 3 days after SE induction. All procedures were approved by the Institutional Animal Care and Use Committee for Dankook University (DKU-14-034).
Tissue processing
The mice were anesthetized with ethyl ether and transcardially perfused with cold saline, followed by 4% paraformaldehyde in phosphate buffered saline (PBS), pH 7.4. Their brains were post-fixed for 4 h, then cryoprotected in 30% sucrose in PBS. Sequential coronal sections (25 µm thick) through the hippocampus were prepared using a cryocut microtome (CM3050S, Leica, Germany).
Cresyl violet staining
Live cells were labeled using cresyl violet. The tissues were mounted on gelatin-coated slides for overnight before use. After dehydration in a graded alcohol series, hippocampal sections were stained for 20 min with pre-warmed 0.3% cresyl violet solution at room temperature. After destaining with a solution of 95% ethanol and 0.3% glacial acetic acid, the sections were dehydrated using 100% ethanol, followed by 100% xylene. The sections were then mounted with DPX.
Fluoro-Jade B staining
Dead or dying cells were labeled using Fluoro-Jade B. The tissues were mounted on gelatin-coated slides for overnight before use. After dehydration in a graded alcohol series, hippocampal sections were incubated in 0.06% potassium permanganate solution for 10 min. Next, the sections were stained with 0.0004% Fluoro-Jade B solution containing 0.1% glacial acetic acid for 20 min at room temperature. They were then washed with distilled water, dried, and mounted with DPX.
Immunohistochemistry
For immunohistochemical labeling, free-floating sections were sufficiently washed with PBS, then blocked with 5% BSA and 0.4% Triton X-100 in PBS. The tissues were then incubated for overnight at 4℃ with mouse anti-glial fibrillary acidic protein antibody (1:500; Millipore, Temecula, CA, USA) or rat anti-CD11b antibody (1:200; Abcam, Cambridge, MA, USA). After washing, the sections were incubated with Alexa Fluor 488 anti-rat IgG (1:500; Life Technologies, Carlsbad, CA, USA) and Alexa Fluor 555 anti-mouse IgG (1:500; Life Technologies, Carlsbad, CA, USA) in PBS for 2 h at room temperature. After washing in PBS, coverslips were inverted onto the slides over a drop of VECTASHIELD Mounting Medium (Vector Laboratories, Inc., Burlingame, CA, USA). Alexa Fluor 488 (excitation, 488 nm; emission, >519 nm) and Alexa Fluor 555 (excitation, 555 nm; emission, >565 nm) -labeled neurons were imaged using confocal microscopy (LSM 700, Carl Zeiss, Germany).
Statistical analysis
Data are expressed as means±SEM. Student's t test was used for statistical comparisons (GraphPad Prism 5 software v5.01, San Diego, CA, USA). A p value of < 0.05 was considered statistically significant.
Go to :
RESULTS
Effect of lithium post-treatment on pilocarpine-induced status epilepticus
To investigate whether lithium could inhibit seizure susceptibility after pilocarpine-induced SE, we employed the pilocarpine model of SE in mice. Eight-week-old, C57BL/6 male mice were injected with atropine (1.2 mg/kg, i.p.), then injected with pilocarpine hydrochloride (320 mg/kg, i.p.) 30 min later. Lithium (80 mg/kg, i.p.) was administered 15 min after the pilocarpine injection. Behavioral changes after the injection with pilocarpine hydrochloride were recorded according to Racine's scale [14] as described in Materials and Methods. Briefly, all mice developed diarrhea, piloerection, and other signs of cholinergic stimulation about 5 min after pilocarpine injection. Mice then, exhibited head bobbing, scratching, chewing, and exploratory behavior during the following 15 to 20 min. Recurrent seizures started around 15 to 20 min after pilocarpine administration. These seizures progressed to SE at about 35 to 40 min after pilocarpine administration [1516]. The mean latency to onset of SE was significantly delayed in the lithium-treated group compared to the vehicle-treated group (vehicle, 28.6±1.6 min; lithium, 36.0±3.9 min) (Table 1). The percentages of seizures and SE were also decreased in the lithium-treated group (vehicle, 96.8%; lithium, 79.3%). Mortality observed over a period of 3 days following pilocarpine hydrochloride injection was reduced in the lithium-treated group (vehicle, 38.7%; lithium, 27.6%). In all of the animals with SE, myoclonus persisted for more than 6 h after pilocarpine injection and no difference in behavioral patterns was observed between the two groups. These data indicate that lithium significantly delayed the onset time of SE and reduced mortality compared to the vehicle-treated group.
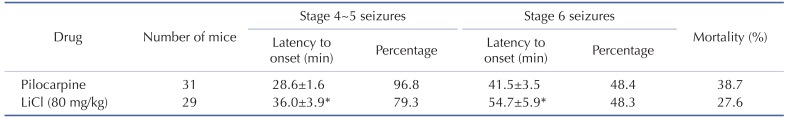
Lithium post-treatment decreased hippocampal damage after SE
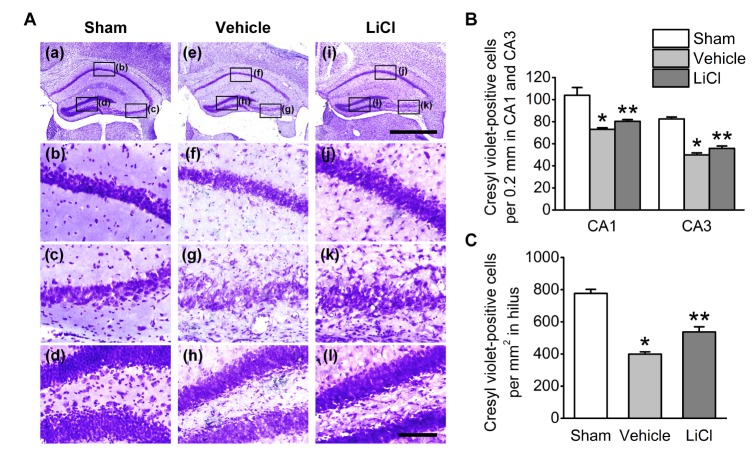
We observed reproducible neuronal death in the hippocampus at 3 days after pilocarpine injection, consistent with a previous study [17]. Neuronal cell death was determined using cresyl violet staining (Fig. 1). Many pyknotic nuclei were observed in the hippocampal CA1, CA3 and the hilus subfields of vehicle-treated mice (Fig. 1A(e)~A(h), 1B and 1C). In the sham-treated mice, cresyl violet-positive cells were detected in CA1 at 103.9±7.0 cells/0.2 mm, followed by CA3 at 82.4±1.8 cells/0.2 mm and the hilus at 776.8±25.0 cells/mm2 (Fig. 1B and 1C). Compared to the sham-operated mice, cresyl violet-positive cells were dramatically reduced in CA1 at 73.1±1.3 cells/0.2 mm, followed by CA3 at 49.9±1.9 cells/0.2 mm and the hilus at 399.2±13.8 cells/mm2 in the vehicle-treated mice (Fig. 1B and 1C). However, cresyl violet-positive cells were significantly increased in CA1 at 80.4±1.5 cells/0.2 mm, CA3 at 55.8±2.1 cells/0.2 mm and the hilus at 537.0±32.1 cells/mm2 in the lithium-treated mice compared to the vehicle-treated mice (Fig. 1B and 1C). In parallel with the cresyl violet staining data, we confirmed that obvious neuronal cell death at 3 days post SE was detected in CA1, CA3 and the hilus (Fig. 2) (vehicle, 23.6±1.5, 7.8±3.1, and 644.9±63.1, respectively). Furthermore, neuronal cell death was significantly decreased in all subfields of the hippocampus in the lithium-treated mice (CA1, 16.8±2.9, CA3, 1.5±0.4, hilus, 442.9±74.9). However, Fluoro-Jade B-positive cells were not detected in the hippocampal regions of the sham-treated mice (data not shown). Quantitative analyses of Fluoro-Jade B-positive cells indicate that lithium post-treatment induced robust neuroprotective effects against pilocarpine-induced neuronal cell death in the hippocampus (Fig. 2B and 2C).
 | Fig. 1Neuroprotective effect of lithium in the mouse hippocampus after pilocarpine-induced SE as evaluated by cresyl violet staining.(A) Representative images of cresyl violet-stained brain coronal sections 3 days after pilocarpine-induced SE. Treatment with lithium chloride (80 mg/kg) protected the CA1, CA3 and hilus cells of the hippocampus 15 min after pilocarpine injections. The scale bars in i and l indicate 1 mm and 100 µm, respectively. (B), (C) Quantitative analysis of neuronal damage in the sham-, vehicle-, and LiCl-treated groups, respectively. *p<0.05 relative to sham. **p<0.05 relative to vehicle.
|
 | Fig. 2Lithium reduces neuronal cell death in the mouse hippocampus after pilocarpine-induced SE.(A) Representative images of Fluoro Jade B-stained brain coronal sections 3 days after pilocarpine-induced SE. Treatment with lithium chloride (80 mg/kg) protected the CA1, CA3 and hilus cells of the hippocampus 15 min after pilocarpine injection. The scale bars indicate 100 µm. (B), (C) Quantitative analysis of neuronal damage in the vehicle- and LiCl-treated groups. *p<0.05 relative to vehicle.
|
Reduced glial reaction in the lithium-treated group
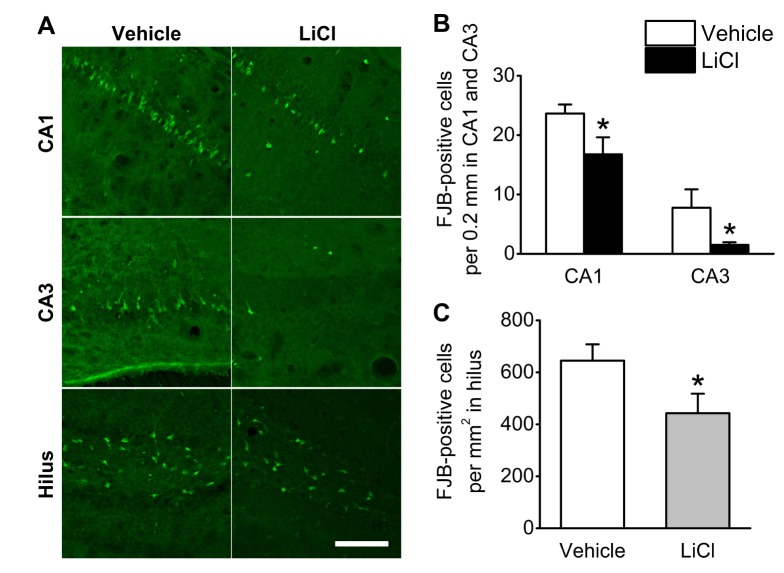
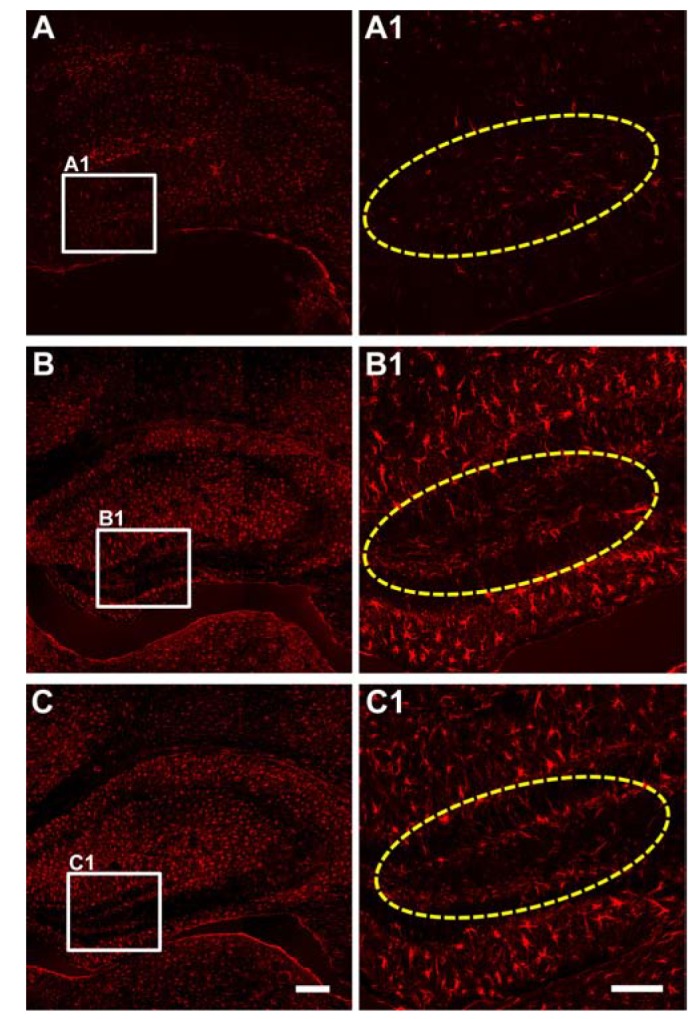
Extensive reactive astrogliosis and microglial activation (microgliosis) have been associated with SE-induced neuro-pathological changes [18]. Therefore, we examined the effects of lithium on astrocytic and microglial changes by testing GFAP and CD11b immunoreactivity (astrocyte and microglia markers, respectively). At 3 days after pilocarpine-induced SE, more GFAP-labeled astrocytes were observed in CA1, CA3 and the hilus (Fig. 3B and 3B1). However, there was no prominent difference in GFAP immunoreactivity between the vehicle- and lithium-treated groups. In addition, CD11b-labeled cells were increased in all three hippocampal subfields 3 days following pilocarpine-induced SE, while no distinct CD11b immunoreactivity was observed in any of the hippocampal subfields of the lithium-treated animals (Fig. 4).
 | Fig. 3Astrocyte activation in the hilus.Representative images of GFAP immunoreactivity from sham-treated animals (A and A1), vehicle-treated animals (B and B1) and LiCl-treated animals (C and C1). Animals were perfused at 3 days after the pilocarpine-induced SE. The scale bars in C and C1 indicate 200 µm and 100 µm, respectively.
|
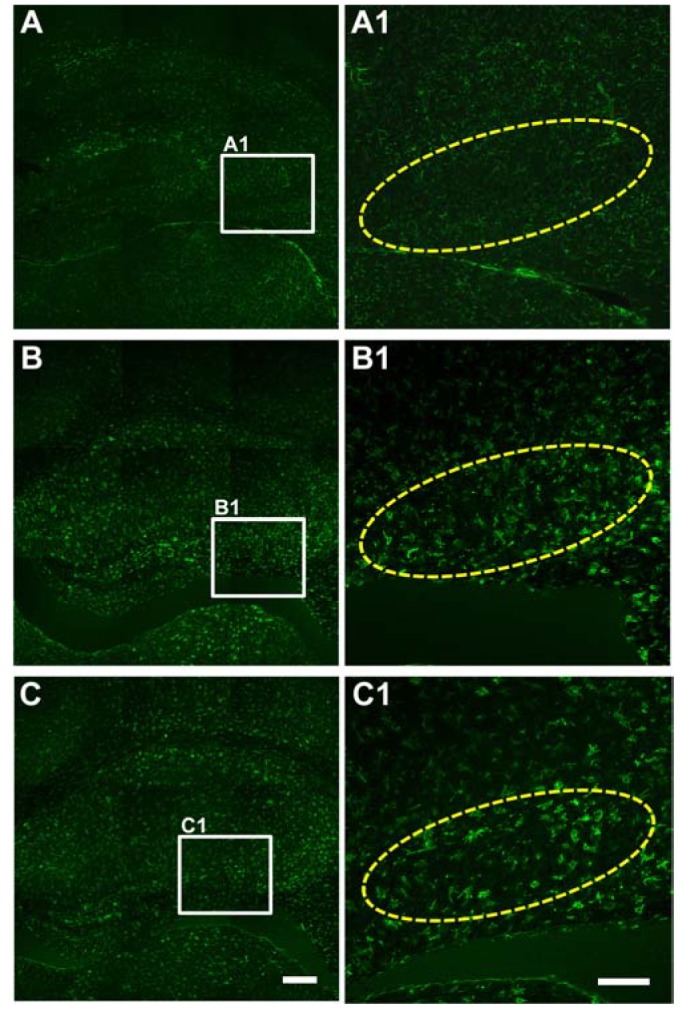 | Fig. 4Microglia activation in CA3.Representative images of immunoreactivity to CD11b, a marker of activated microglia, from sham-treated animals (A and A1), vehicle-treated animals (B and B1) and LiCl-treated animals (C and C1). Animals were perfused at 3 days after the pilocarpine-induced SE. The scale bars in C and C1 indicate 200 µm and 100 µm, respectively.
|
Go to :
DISCUSSION
This study was designed to investigate the effects of lithium on seizure susceptibility and hippocampal damages following pilocarpine-induced SE in mice. The data presented here suggest that lithium post-treatment reduces seizure susceptibility and mortality as well as neuronal death without exerting a pronounced effect on glial activation in the hippocampus.
The most intriguing finding of our study was that lithium post-treatment alleviated seizure susceptibility and mortality. Our data agree with those of other studies on the antiepileptic activity of lithium. In a pentylenetetrazol-induced convulsion model, lithium possessed anticonvulsant activity [56]. After seizures were induced by electroshock, lithium chloride at high concentration caused a significant decrease in seizure susceptibility [7]. Schmidt [8] also reported anticonvulsant activity of lithium in a pentylenetetrazol-kindled rat model. In clinical studies, lithium has produced anticonvulsant activity in patients with temporal lobe epilepsy [192021]. It is well-documented that increased intracellular Ca2+ concentration plays a significant role in epileptic activity [2223], leading to neuronal death in SE [24]. Glutamate receptors are likely involved in modulating susceptibility to seizure development [2526]. Furthermore, lithium attenuates NMDA-mediated Ca2+ influx into neurons [27]. Taken together, these data suggest that lithium could reduce seizure susceptibility, at least in part by the influx of Ca2+ through the NMDA receptors involved in anticonvulsant activities.
In our study, acute administration of lithium at the dose of 80 mg/kg, i.p. reduced the seizure susceptibility significantly (Table 1), which is in line with other study reporting the anticonvulsive effect of lithium in pentylenetetrazole seizure model [28]. However, Ghasemi et al. have demonstrated that acute administration of lithium at doses >50 mg/kg had no significant effect on pentylenetetrazole-induced clonic seizure threshold [29]. This difference might be related to the different animal species or the different models of seizure used in the study. There are some reports demonstrating that acute administration of lithium significantly decreased seizure susceptibility at similar or even higher doses [728]. In addition, we intraperitoneally injected mice with lithium at 15 min after pilocarpine injection, which is likely to work at the time of induction of SE after pilocarpine injection [1516]. Indeed, other studies have shown that when lithium was administered by intraperitoneal injection, the action of lithium was significant after about 30 min after treatment [282930].
Neuronal loss in the hippocampal subfields is a common neuropathological feature of temporal lobe epilepsy. The locations of neuronal loss in pilocarpine-induced epileptic mice observed here are in accordance with previous studies [31]. The timing of neuronal death induced by pilocarpine was distinguishable depending on the location of the lesions. Consistent with previous studies [31], neuronal loss was seen in all hippocampal subfields at 3 days after SE. In the present study, we generated data from cresyl violet staining and Fluoro-Jade B staining, indicating lithium's neuroprotective effects against pilocarpine-induced SE. Compared with the vehicle-treated epileptic mice, lithium induced pronounced neuroprotection at 3 days after SE throughout all hippocampal subfields. Our data are consistent with other studies on the neuroprotective effects of lithium. Lithium has been shown to reduce neuronal death in several neurodegenerative disease models, including Parkinson's disease [3233], tauopathies [34], Huntington's disease [35], amyotrophic lateral sclerosis [36] and multiple sclerosis [37]. Although the mechanisms underlying the neuroprotective effects of lithium are not clear, modulation of inflammatory activity, glutamate receptors, GSK-3, and inositol depletion [3338] could contribute to this phenomenon.
The neuronal damage induced by SE can be accompanied by glial activation, which leads to the progression of neurodegeneration [31]. The inflammatory response is mediated by activated microglia, which is considered an important strategy in the treatment of neurodegenerative diseases. Shapiro et al. [39] showed that microglial activation was observed in all three hippocampal regions at 1 and 2 days after seizure onset and continued for 5 days after onset, although significantly greater microglial activation was observed in the hilus beginning at day 1. In the present study, our data showed that astrocyte and microglial activation was detected in all hippocampal subfields at 3 days after SE. Interestingly, we could not detect any pronounced differences in glial activation between the vehicle- and the lithium-treated mice following SE, although lithium treatment decreased seizure susceptibility and mortality, as well as neuronal death following pilocarpine-induced SE. There may be mechanisms, other than modulation of the inflammatory response underlying the neuroprotective actions of lithium against pilocarpine-induced SE.
In summary, lithium significantly reduced seizure susceptibility and mortality as well as neuronal death following pilocarpine-induced SE in the hippocampus, while showing no prominent effect on glial activation after SE. Lithium therefore has strong potential to provide beneficial effects for patients with
SE.
Go to :
 XML Download
XML Download