

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Understanding absorption, distribution, metabolism, excretion (ADME) as well as pharmacokinetics (PK) is critical in drug discovery and development. This approach typically begins early in the drug discovery phase, when hit compounds are available. As the project moves forward to the lead optimization phase, ADME/PK contributes significantly in rank-ordering compounds for further testing, aids in the dose selection in nonclinical in vivo pharmacology studies, establishes structure-activity relationships (SAR) for structural modifications to improve the physicochemical and ADME properties and eventually supports candidate selection [1]. Once a candidate is selected, human PK prediction can also be conducted to estimate the dose-dependent drug exposure and its anticipated pharmacological response as well as the potential toxicological effects or even the potential drug-drug interactions.
Drug-drug interactions (DDIs) have led to contraindications, withdrawal from the market and non-approval of drugs by regulatory agencies [2]. Therefore, prediction for a potential risk of DDIs between the candidate drug and the co-administered drugs would be very important within the pharmaceutical industry in order to improve the safety and success rate of new drugs. Early prediction of DDIs has also become useful to support clinical candidate selection. DDI studies and their interpretation have been described in a white paper from the Pharmaceutical Research and Manufacturers of America (PhRMA) [3] and in a guidance document from the US FDA.
Over the last decade, model-based PK/DDI prediction using in vitro data has been increased significantly [45]. Initially, simple empirical static models were applied to evaluate the DDI risk to predict steady state DDI [6789]. However, these static models are often restricted when predicting the ratio of substrate exposures before and after perpetrator treatment and are unable to simulate dynamic state DDI. Therefore, a dynamic DDI modeling and simulation has been proposed to predict DDI which can represent in vivo DDI more realistically [10]. Recently, the development of physiologically-based pharmacokinetic (PBPK) models has extended to the DDI evaluation of dynamic approaches with the ability to simulate time-varying substrate and perpetrator kinetics at interaction sites [111213]. In addition, commercial programs (e.g. GastroPlus®, Simcyp®) dedicated to DDI predictions are also introduced which could combine both in vitro/in vivo data and clinical study results to help the evaluation of dynamic DDIs more efficiently. The PBPK model is typically composed of specific compartments corresponding to different body tissues and these tissues were also connected by the circulating blood system. Each compartment is defined by a tissue-specific volume, blood perfusion rate, enzyme/transporter expression levels and tissue-plasma partition coefficient (Kp) etc [13].
Herein, we describe a case study using caffeine and ciprofloxacin to illustrate the use of GastroPlus in predicting PK and DDI liability using in silico, in vivo and in vitro derived parameters. This case is composed of five processes such as (1) simulation, (2) verification, (3) understanding of parameter sensitivity, (4) optimization of the parameter and (5) final evaluation. The objective of this study is to demonstrate the application of in silico tools for simulation, process for understanding of drug parameter sensitivity, optimization of drug parameter, evaluation of PBPK model to provide a satisfactory prediction of PK profile and DDI result. However, it should be emphasized that these simulations are quite dependent on the quality of the input parameters and mechanistic understanding of the processes driving PK. A number of more detailed case studies using in silico program are also available in the literature illustrating the value of such modeling and simulation techniques [1415161718].
Go to : 
METHODS
PBPK modeling program
All modelings were performed using GastroPlus (Version 9.0). The physicochemical and ADME properties of compounds were estimated and optimized by the ADMET predictor module in GastroPlus.
The Advanced Compartmental Absorption Transit (ACAT) model was used to predict the rate and extent of oral absorption in human. The ACAT model is based on the Compartmental Absorption Transit (CAT) model described by Yu and Amidon [19]. In short, this model is a physiologically based transit model which describes the dissolution, uptake and absorption of a compound as it transits through the different segments of the digestive tract. Several in vitro and in silico input data, e.g. solubility, permeability, logP, particle size, acid dissociation constant (pKa) etc with a series of differential equations were also used to model the kinetics associated with each of these processes. Each organ was also assumed to be perfusion rate limited, and the liver and kidney were considered to be the only organs to eliminate the compounds in this study.
Volume of distribution at steady-state (Vss) was estimated using the predicted tissue-to-plasma partition coefficient (Ptp) [20]. The predicted Ptp values for each tissue were obtained from drug-specific physicochemical parameters using the tissue composition-based equation derived from Poulin and Theil [21]. These equations assume that the compound distributes homogenously into the tissue and plasma by passive diffusion and accounts for both nonspecific binding to lipids and plasma proteins estimated by lipophilicity and plasma protein binding (PPB), respectively.
Compounds and PBPK modeling
The physicochemical properties of caffeine and ciprofloxacin are summarized in Table 1. The workflow for PBPK modeling is also presented in Fig. 1. Firstly, a PBPK model for each compound was built in human. And then, the model was compared with the observed PK data published in the literature. If the PBPK model data were not close enough to the observed data, the parameter sensitivity analysis (PSA) was performed to understand the root cause of discrepancy between the observed PK data and the simulated PBPK data until the root cause of discrepancy is discovered. Once the simulated PBPK model was verified, it was re-evaluated by other new PK data to confirm its predictability. Finally the DDI prediction was performed and evaluated using the confirmed PBPK models. References for verification and evaluation of PBPK model were summarized in Table 2.
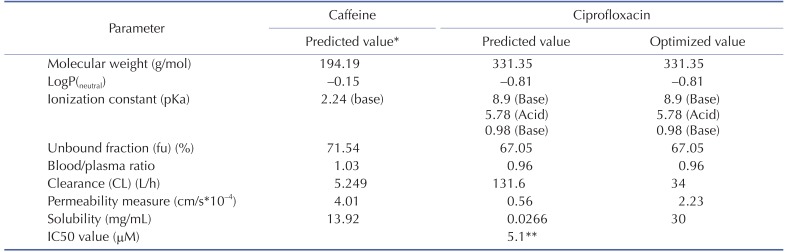
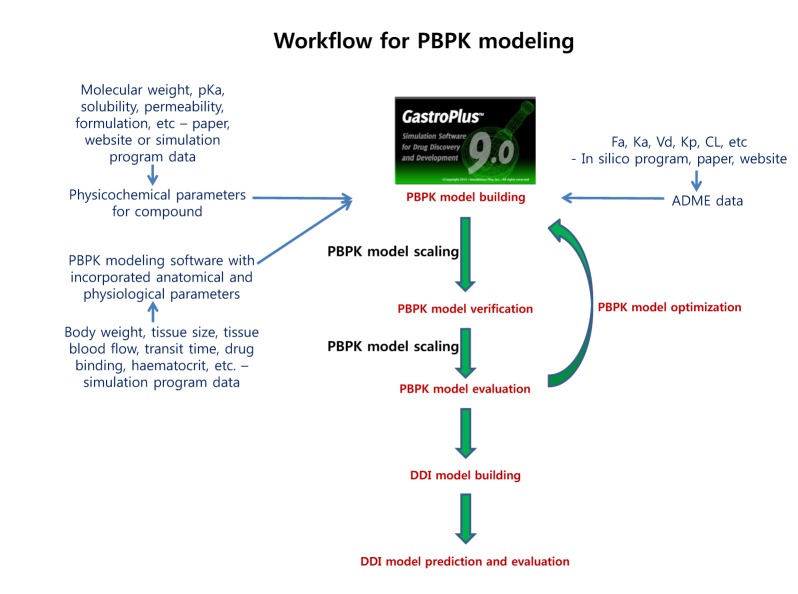
Table 1
Summary of caffeine and ciprofloxacin input parameters in GastroPlus
*All of caffeine input parameters were the predicted values and weren't optimized.
**In vitro IC50 value on the CYP1A2 measured by Zhang et al was used [29].
![]()
The physicochemical and ADME properties of caffeine such as pKa, logP, unbound fraction, permeability, solubility, clearance (CL) by liver (or kidney), and blood/plasma ratio etc were estimated by the ADMET predictor module in GastroPlus. The PBPK modeling of caffeine was performed as mentioned above. The physicochemical and ADME properties of ciprofloxacin were also estimated by ADMET predictor module in GastroPlus and PBPK modeling was conducted as mentioned above.
The PBPK model was assessed in terms of the fold error between the observed values and the predicted values. The following statistics were used to assess the prediction accuracy.
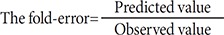
The R2 statistic (square of the Pearson product moment correlation coefficient) was used to quantify the extent of linear relationship between predicted and observed parameters. In this study, two-fold error would be an acceptable prediction which is also widely applied within pharmaceutical industries [15222324].
DDI modeling and simulation
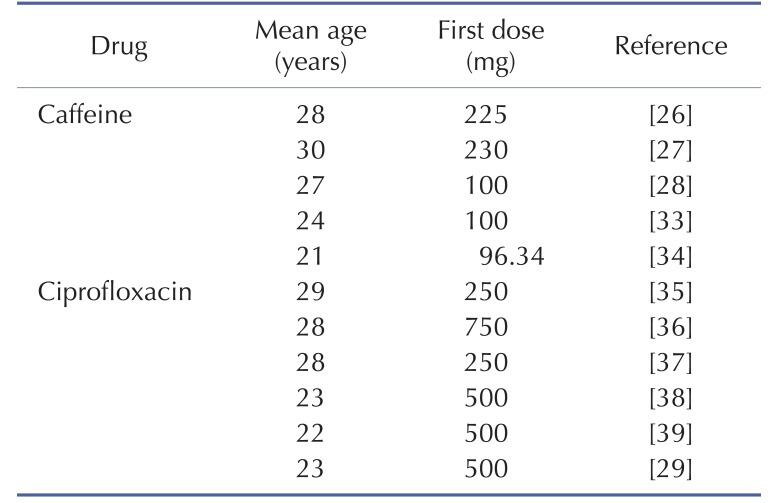
DDI modeling for caffeine and ciprofloxacin was conducted using the dynamic simulation in DDI module of GastroPlus. The verified PBPK models were used for DDI prediction by considering the inhibitory effect of ciprofloxacin as a perpetrator on caffeine. Caffeine was assumed to be exclusively metabolized by the metabolic enzyme CYP1A2 without significant involvement of any other drug transporters or metabolic enzymes in this DDI simulation. Although several clinical PK studies for caffeine or ciprofloxacin had been previously reported, the full concentration-time profile data by DDI between caffeine and ciprofloxacin were quite limited. In this study, PK results from various clinical studies were used in the DDI modeling and simulation for caffeine and ciprofloxacin [252627]. The detailed model input parameters were also listed in Table 1. The in vitro IC50 value on the CYP1A2 measured by Zhang et al was also used for this DDI simulation [28]. Population mean age, dose, dose interval and the duration of administration of ciprofloxacin and caffeine were determined according to the regimen of the clinical DDI studies. The detail values are listed in Table 3.

Go to :
RESULTS
PBPK model for caffeine
The workflow for building, verifying and evaluating PBPK model for caffeine and ciprofloxacin is described in Fig. 1. The physicochemical and ADME properties of caffeine and ciprofloxacin were predicted using ADMET predictor module in GastroPlus and are summarized in Table 1. The PBPK model in human was built for oral administration (PO) dosing using human physiological PBPK model in GastroPlus. The comparison between the predicted value and the observed value was presented in Fig. 2. The simulated PK parameters and concentrations were within 2-fold error of the observed PK parameters (Cmax, Tmax, AUClast) when the predicted ADME parameters by ADMET predictor were used. The predicted results were comparable to those from observed results.
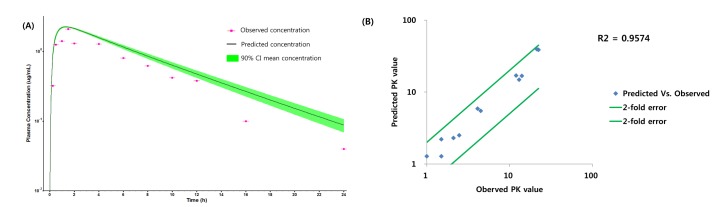
PBPK model for ciprofloxacin
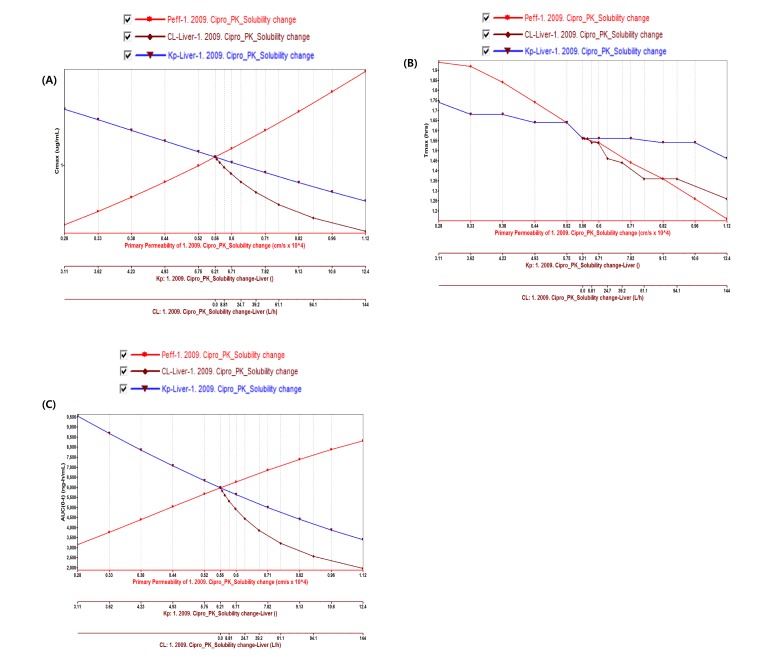
PBPK model for ciprofloxacin was also built using the method described in the experimental section. The physicochemical properties and ADME properties of ciprofloxacin were also predicted by the ADMET predictor module in GastroPlus and are summarized in Table 1. The PBPK model for ciprofloxacin in human was built for PO dosing using human physiological PBPK model in GastroPlus. The verification and optimization results of PBPK modeling are presented in Fig. 3. When verification was conducted using the predicted input value only based on the structure of ciprofloxacin, the PK parameters such as Cmax, Tmax and AUClast values were much lower than the observed value (Fig. 3A). Several factors were considered as a root cause of the discrepancy and the first one to be investigated was solubility. The ciprofloxacin used in clinical PK studies was a salt form, ciprofloxacin HCl. Salt formation offers many advantages to pharmaceutical products as it can improve the solubility, dissolution rate, permeability and efficacy of the drug. However, the ciprofloxacin used for PBPK simulation was a free-base form. This difference could contribute big difference in terms of absorption rate. In addition, the predicted and measured value of solubility were quite different. Therefore, the solubility input parameter was modified from 0.0266 mg/mL(predicted value by GastroPlus) to 30 mg/mL(value from the Drug Bank). After changing the solubility, the Cmax, Tmax and AUClast values were much more improved than the previous values, however, still lower than the observed values (Fig. 3B). To optimize other properties for PBPK modeling, the parameter sensitivity analysis (PSA) was conducted to predict the impact of the permeability, first pass effect of gut (or oral), fraction unbound in enterocytes, logD, Kp of kidney (and/or liver), and CL of ciprofloxacin on oral exposure. The results of PSA is presented in Fig. 4. The presented results show parameters with significant effects on the oral exposure of ciprofloxacin. The results for parameters having no significant effect on the oral exposure of ciprofloxacin are not shown. Based on the PSA results, little impact on the ciprofloxacin oral exposure was observed by first pass effect of gut or oral, fraction unbound in enterocytes, logD and Kp of kidney (data not shown). However, dramatic changes was observed by permeability, Kp of liver and the CL of ciprofloxacin (Fig. 4). After the parameter optimization for permeability, Kp of liver and CL of ciprofloxacin, the simulated PK values and concentrations were within 2-fold error of the observed PK values (Cmax, Tmax, AUClast) and the concentrations were also well fit for ciprofloxacin (Fig. 3C). In addition, the simulated PK values were also within 2-fold error of the observed PK values from other five different clinical PK studies (Fig. 5). Only one study by Valizadeh et al. [29] showed that the predicted results (Cmax and Tmax) were out of 2-fold error of the observed PK parameters. However, the study by Valizadeh et al. [29] also showed much larger deviation possibly due to significant inter-individual variability in its clinical study and therefore, this paper was excluded from the PBPK modeling.
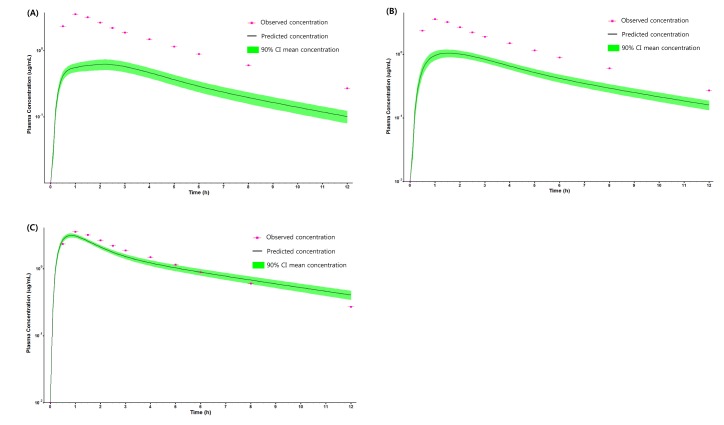 | Fig. 3PK profile of ciprofloxacin after 500 mg PO dose in human.(A) PK profile predicted by only predicted input value, (B) PK profile predicted after changing the solubility of ciprofloxacin, (C) PK profile predicted after optimizing the permeability, Kp of liver and CL of ciprofloxacin.
|
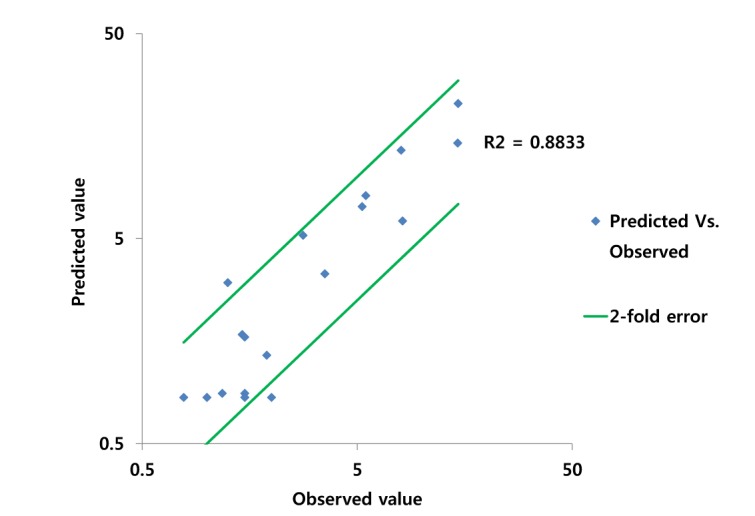
DDI modeling and simulation for caffeine and ciprofloxacin
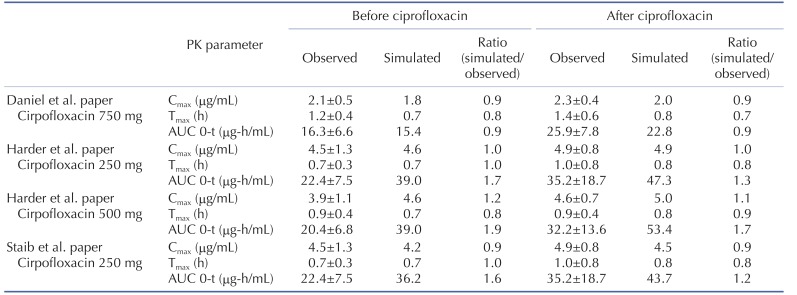
The effect of ciprofloxacin after multiple dose on the PK of caffeine was simulated. The DDI simulations in the presence of CYP1A2 inhibitor ciprofloxacin resulted in various results for Cmax, Tmax, AUClast and the ratio between the observed and predicted PK value (Table 4). As expected, ciprofloxacin was predicted to have an effect on caffeine PK due to CYP1A2 inhibition of ciprofloxacin. With Daniel' paper (ciprofloxacin, 750 mg), the DDI model predicted a mean value of 10% and 50% increase in terms of Cmax and AUC of caffeine compared to the PK result of caffeine alone. Regarding Harder's paper (ciprofloxacin, 250 mg), the DDI model predicted a mean value of 10% and 20% increase in the Cmax and AUC of caffeine compared to the PK result of caffeine before DDI. However, the DDI model predicted a mean value of 10% and 40% increase in the Cmax and AUC of caffeine compared to PK result of caffeine before DDI when the higher dose (500 mg) of ciprofloxacin was co-administered. Staib's paper (ciprofloxacin, QD, 250 mg) also suggested that the DDI model predicted a mean value of 10% and 20% increase in the Cmax and AUC of caffeine compared to the PK result of caffeine before DDI. All of the simulated PK parameters and the rate of change after multiple dosing ciprofloxacin were within 2-fold error of the observed PK parameters and the rate of change after multiple dosing ciprofloxacin. Based on DDI results, the increase of Cmax was insignificant and similar in all of cases. However, the increase of AUC was proportional to the administered dose. In cases of DDI between caffeine and ciprofloxacin, the administered dose was the important factor that could significantly influence the AUC. Also, inter-individual variability on PK parameters was not likely significant in this study. Overall, the predicted results were comparable to those from observed results. The verified PBPK model using in vivo PK data could be used to predict in vivo DDI result.
Go to :
DISCUSSION
The prediction of PK profile using compartmental PK models, as traditional PK model, is convenient; however, extrapolation and assumptions about distributional kinetics are essential [30]. On the contrary, the PBPK models have been demonstrated to facilitate cross species extrapolation and more accurately predict the PK profiles [31]. This PBPK approach is particularly useful when the DMPK resource is limited and unable to conduct many in vivo PK studies on a regular basis at the discovery stage. Once PBPK model building and verification in one preclinical species are performed, the PBPK model can be scaled or applied to other species or even to human. Regardless of a type of the PK model used, this approach for predicting PK profile has a broad application; such as (1) determining in vitro properties required to obtain the target PK profile, (2) rank-ordering compounds in discovery most likely for the desired exposure profile in animals or human, (3) assessing the effect of food on absorption in human, and (4) determine attributes governing bioavailability and required to obtain a desired profile [1]. This approach is also very helpful even in roughly-predicting DDI-liabilities at the early drug discovery. GastroPlus has a quantitative structure-activity relationship (QSAR) model function (ADMET predictor) and can estimate a number of ADMET properties of new chemical entities based on their molecular structures.
This study described herein is an example of where in silico programs such as PBPK and ADMET predictor have been able to utilize the parameter estimates to adequately predict the observed clinical PK profile and DDI result or optimize the PK model parameters with comparison and interpretation between predicted and observed results, using PSA. Based on the simulation, the DDI liability of ciprofloxacin as a perpetrator on caffeine appears to be minimal and no significant increase of Cmax and AUC in caffeine was predicted. In general, the more in silico derived parameters are used as inputs for the model, the more assumptions are required. Obviously the most closely arrayed parameter set such as in vivo measured values will likely provide the best prediction in modeling and simulation. Therefore, when the more in silico derived parameters are used in PBPK modeling, the more caution should be taken to make sure that the result is acceptable and the parameters could suitably influence the result. Therefore, a good understanding of the physicochemical and ADME properties of the drug and its role in oral exposure could be critical in any modeling and simulation exercise. Once correlations are established, in silico derived parameters could be used as a powerful tool to predict PK profile as well as DDI liability for the new chemical entities in the early drug discovery and development phase.
Go to :
CONCLUSION
Predicting potential drug–drug interactions is one of the main areas where PBPK approaches have offered significant advances in recent years. As a recommended practice, the verification of an appropriate PBPK model with the parameter sensitivity analysis as well as in vivo data could provide the increased confidence in DDI simulation outcome. This approach could be particularly useful in identifying potential key factors that could lead to significant impact on the extent of human exposure and DDI. This case study suggested that in silico derived parameters could be useful and acceptable for PK prediction if carefully applied. This study also suggested that the optimization module such as PSA could be helpful for optimizing important parameters for the prediction of human exposure and DDI simulation.
Go to :
 XML Download
XML Download