

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Caspases are a family of cysteine proteases which are ubiquitously expressed and play a significant role in apoptosis and autophagy [12]. Activation of caspases generally induces selective cleavage of specific target proteins, leading to the inactivation and degradation of the target proteins [12].
Death caspase 1 (DCP1) is a Drosophila caspase gene which cleaves a cysteine protease substrate and is important in development and oogenesis [2]. Dcp-1 deletion mutants show a defect in cell death in the germ line during mid-stage oogenesis under conditions of nutrient deprivation [134], while a cell death phenotype in normal flies is observed during mid-stage oogenesis under nutrient deprivation [156]. Over-expression of a single copy of the truncated N-terminal region of Dcp-1 (constitutively active Dcp-1) in the eye results in a slightly rougher and less pigmented eye [4]. Recent studies in mammals and flies suggest that caspases also have a significant role in non-apoptotic cell death (such as autophagy) as well as in apoptosis [78].
Autophagy is involved in cell survival as well as cell death [39]. When cells lack essential nutrients, the autophagy pathway is activated in order to provide nutritional components [10]. Under certain environmental conditions, autophagy is also able to induce autophagic cell death [10]. The diverse biological roles of autophagy affect aging, development, and tumorigenesis [311]. Levels of autophagy are decreased by activation of the PI3K, Akt, and mTOR signaling pathways in cancer cells, whereas phosphatase and tensin homolog (PTEN), tuberous sclerosis 1 (TSC1), tuberous sclerosis 2 (TSC2), autophagy-specific gene 4 (Atg4), and beclin 1, which are known to be tumor suppressors, induce autophagy [912131415].
p53 is a well-known tumor suppressor, which is known to cause cell cycle arrest, autophagy, and apoptosis [161718]. The function of p53 in cell cycle arrest and apoptosis involves the transcriptional activation of proteins such as p21, p27, Puma, Noxa, and Bax [181920]. Genotoxic, hypoxic, and oncogenic stressors can induce transcriptional activation of p53 targets as well as cytosolic accumulation of p53 protein with mitochondrial localization, leading to rapid mitochondrial apoptosis [2021]. Moreover, p53 has recently been identified as a regulator of autophagy. Autophagy can be induced in a p53-dependent manner in response to genotoxins [21]. One of the mechanisms of p53-induced autophagy involves activation of AMPK kinase with subsequent activation of TSC1 and TSC2 kinases, which leads to the inhibition of mTOR kinase, which controls the translational machinery and cell growth [19]. Activation of p53 also leads to upregulation of PTEN and TSC2 transcription, which may contribute to the long-term suppression of mTOR [222324]. Another mechanism of p53-induced autophagy is through transcriptional activation of DRAM, a p53 target and a lysosomal protein [1825]. Autophagy induced by p53 may facilitate cell cycle arrest and is known to mediate the selective degradation of damaged molecules and organelles in order to provide an energy source for the damage repair process and promote cell healing [172021].
The Drosophila system is an attractive model to study the molecular mechanisms of cellular phenotypes, including apoptosis and autophagy, because of the organism's short generation time and well-established, simple screening methods [14]. Our previous study identified 72 genes that regulate cell death in Drosophila , via modifier screening of death caspase 1 (DCP1) [1]. Although a gene may be known to regulate a phenotype in a certain species or tissue type, it can act in other species or tissues in much different ways. Also, some genes, including Alfy and Tak1, were previously known to induce autophagy; however, their correlations with p53 activation have not yet been reported. In this study, 16 genes related to caspases in Drosophila were tested in human cell lines to assess whether they can induce apoptosis or autophagy through the p53 pathway.
METHODS
Cell culture and transfection
HEK 293T cells, HEK 293A cells, MCF7 and U2OS human osteosarcoma cells were grown in Dulbecco's Modified Eagle's Medium (DMEM; Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; HyClone, Logan, UT, USA), 100 IU/ml penicillin, and 100 µg/ml streptomycin (Gibco-BRL). All cells were maintained at 37℃ in a humidified incubator with 5% CO2. HEK 293T cells were transfected as described in the figure legends, using the FuGENE HD Transfection Reagent (Roche, Mannheim, Germany), according to the manufacturers' protocol.
Immunoblot analysis
The GFP-LC3 plasmid was a kind gift from Dr. Tamotsu Yoshimori (Osaka University, Osaka, Japan). Forty-eight hours after transfection, whole-cell protein lysates were prepared from transfected cells using 5X sample buffer (250 mM Tris-HCl, pH 6.8, 10% SDS, 30% glycerol, 0.5 M DTT, and 0.02% bromophenol blue). Denatured protein (20 µg) was loaded on 8~12% gels, and electrophoresis was performed. The gels were then transferred onto nitrocellulose membranes for 2 h at 4℃. Following transfer, the membranes were blocked for 1 h at room temperature in blocking buffer (5% skim milk in PBS). The membranes were then incubated overnight at 4℃ with primary antibodies diluted in 1X PBST buffer. The following primary antibodies were used: GFP (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:1,000 dilution), and β-actin (Sigma-Aldrich, 1:10,000 dilution). The membranes were washed three times with PBST. Secondary antibodies were diluted in PBST and were added for 1 h 40 min at room temperature. The following secondary antibodies were used: anti-rabbit IgG HRP-linked antibody (Cell Signaling Technology, 1:2,000 dilution) and goat anti-mouse IgG-HRP conjugated antibody (Chemicon, 1:5,000 dilution). The membranes were washed five times with PBST. Chemiluminescent detection was accomplished using SuperSignal West Pico substrate (Pierce, Rockford, IL, USA), and the images were analyzed with an LAS 4000 instrument (Fujifilm).
RT-PCR
For each sample, total RNA from cells was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Reverse transcription was carried out with the Transcriptor First Strand cDNA synthesis Kit (Roche) using the provided protocol. PCR was then performed as follows: 5 min denaturation at 95℃, followed by 30 cycles of amplification (95℃ for 1 min, 55℃ for 1 min, and 72℃ for 2 min), and an additional extension for 10 min at 72℃. RT-PCR was performed using a GeneAmp PCR System 9700 (Applied Biosystems, CA, USA).
Evaluation of caspase activity
Caspase activity was determined using the Caspase-Glo 3/7 Assay (Promega, Madison, WI, USA). Forty-eight hours after transfection, the cells were harvested. Cells were seeded at a density of 8,000 cells/well in a 96-well plate and mixed with the Caspase Glo 3/7 reagent (1:1 ratio). Samples were incubated for 1 h at room temperature. The luminescence of each sample was measured in a Wallac Victor 1420 multilabel counter (PerkinElmer, Waltham, MA, USA).
Determination of autophagy or apoptosis using GFP-LC3
For determining autophagy, cells were seeded at a density of 1.5×105 cells/well in 12-well plates the day before transfection, and 50 ng of GFP-LC3 along with 450 ng of either TAK1 or control vector was co-transfected into the indicated types of cells using FuGENE HD Transfection Reagent (Roche). Forty-eight hours after transfection, GFP-LC3 puncta were visualized using a Leica DM6000B microscope. Cell death was assessed using a LDH release assay (Cytotoxicity Detection KitPLUS LDH, Roche). The mTOR plasmid was purchased from Addgene (Cambridge, MA, USA), and the Bax plasmid was obtained from KRIBB (Daejeon, Republic of Korea). HEK 293T cells were seeded at a density of 7.5×104 cells/well in 24-well plates the day before transfection. The cells were treated with 2 mM 3-MA (Sigma-Aldrich) or 20 nM bafilomycin A1 (Upstate, Billerica, MA, USA) for 1 h before transfection. After 48 h of incubation, a trypan blue (Welgene, Daegu, South Korea) exclusion assay was performed to measure cell viability.
Luciferase reporter assay
For the luciferase reporter assay, pRGC-Luc [26] or p21-Luc [27] reporter plasmid, with or without p53, Mdm2 or the 16 genes identified, was transfected into cells using LipofectAMINE Plus (GIBCO-BRL, CA, USA), in accordance with the supplier's instructions. One microgram of reporter plasmid DNA was mixed in all samples, along with 100 ng of CMV-Ren plasmid (Promega, Madison, WI, USA) as an internal control; this was cotransfected into cells in 6-well plates (Corning Inc., Corning, NY, USA). Luciferase assays were carried out 48 h after transfection, using a Luciferase Assay Reagent kit (Promega, Madison, WI, USA) and a Lumat LB9507 luminometer (Berthold, Bad Wildbad, Germany) [26]. The values obtained were normalized with Renilla luciferase activity.
RESULTS
Identification of autophagy/apoptosis-inducing genes
To identify genes that regulate cell death in Drosophila, we previously screened 15,000 enhancer-promoter (EP) lines and identified 72 DCP1-interacting genes [1], which showed rescued eye phenotype, more severe eye phenotype, or lethal eye phenotype, induced by DCP1 overexpression. In general, DCP1 overexpression aggravated the adult eye phenotype, whereas the co-expression of the 72 Drosophila genes with DCP-1 either rescued the eye phenotype or made it more severe, sometimes leading to lethality. However, the inherent functions of the DCP1-interacting genes, such as apoptosis and autophagy, are poorly understood. In order to reveal the functional characteristics of those genes, we obtained 16 full-length human genes and tested their impact on apoptosis and autophagy in several human cell lines. The detection of green fluorescent protein (GFP)-tagged microtubule-associated protein 1 light chain 3 (GFP-LC3) using fluorescence microscopy is one of the most useful methods to monitor autophagic activity [28]. To investigate whether the 16 genes induce autophagy in vitro, we co-transfected GFP-LC3 with each of the 16 genes or a control vector in several mammalian cell lines, including HeLa, HEK 293A, and MCF7 cells, and examined the accumulation of GFP-LC3 punctate structures (which represent autophagosomes) using fluorescence microscopy. During autophagy, LC3-II localizes to the autophagosomal membrane. Thus, the accumulation of GFP-LC3 puncta provides an effective method of detecting autophagosomes. In control cells, GFP-LC3 punctate structures were not detected, but these structures were markedly increased in cells overexpressing several of the proteins (Fig. 1B, Supplementary Fig. 1 and Table 1).
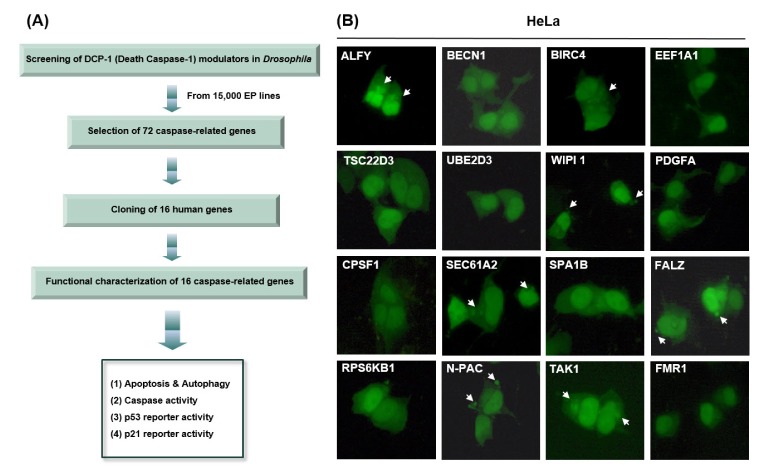
Fig. 1
Overexpression of caspase-related genes induces apoptosis or autophagy.
(A) Screening and selection of 16 cell death-related genes. (B) GFP-LC3 was co-transfected with the indicated genes. After transfection, the cells were incubated for 48 h, and cell death types were then detected using fluorescence microscopy.
![]()
In general, apoptosis appears to be a consequence of mitochondrial membrane permeabilization (MMP), along with a self-amplifying caspase cascade [28]. Cleavage of substrates by apoptotic proteases and activated nucleases induces the characteristic phenotypes of apoptotic cells, such as nuclear condensation and pyknosis, followed by DNA fragmentation [28]. Also, reactive oxygen species (ROS) are produced and lysosomal membrane permeabilization (LMP) occurs [28]. Apoptotic bodies and nuclear condensation were observed in cells overexpressing several of the proteins (Fig. 1B, Supplementary Fig. 1, and Supplementary Table 1).
Fig. 1B shows that ALFY, BIRC4, and TAK1 induced autophagy in HeLa, HEK 293A, and/or MCF7 cells, whereas EEF1A1 induced autophagy only in the MCF7 cell line (Fig. 1B, Supplementary Fig. 1, and Supplementary Table 1). SEC61A2, N-PAC, BIRC4, WIPI1, and FALZ increased apoptotic cell death in HeLa and/or HEK 293A cells. BIRC4 induced autophagy as well as an apoptotic phenotype in HeLa cells. These data suggest that BIRC4 is involved in in both signaling pathways, and that autophagy and apoptosis phenotypes can be induced in a cell type-specific manner by overexpression of DCP1-related genes.
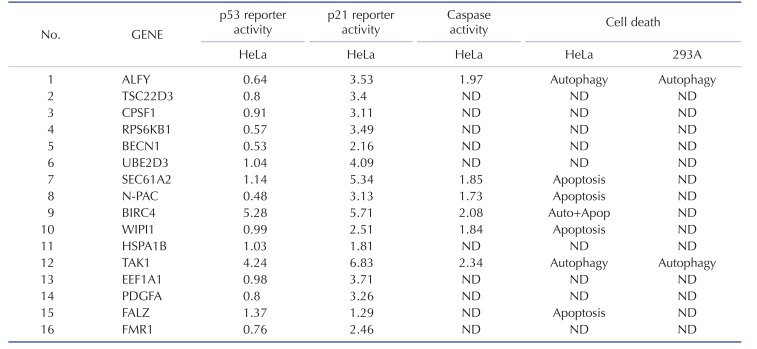
Since caspases have a significant role in the apoptotic and autophagic pathways, we investigated the effect of 6 selected genes with apoptotic and/or autophagic phenotypes on caspase activity. Fig. 2A shows that increased levels of caspase activity were observed when ALFY, SEC61A2, N-PAC, BIRC4, WIPI1, and TAK1 were overexpressed in HEK 293A cells. p53 and Bax, which are known to induce caspase activity, showed similar levels of caspase activity corresponding to their expression, suggesting that apoptosis and autophagy mediated by these genes involve the caspase cascade.
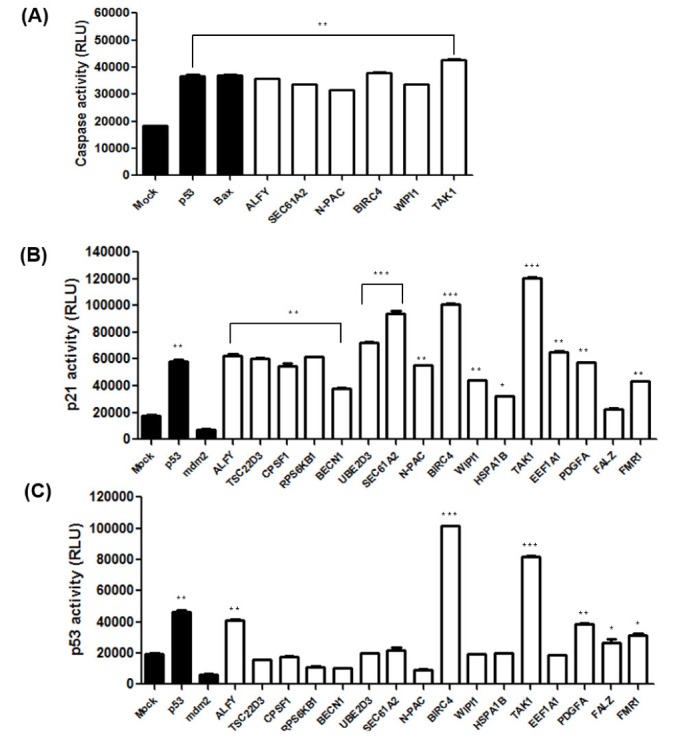
Fig. 2
The effects of caspase-related genes on apoptosis and p53 signaling.
(A) HeLa cells were transfected with the indicated constructs. 48 hours after transfection, cells were harvested and luminescence was measured using the caspase-glo 3/7 assay. (B) p21-luc reporter and the indicated genes were transfected into HeLa cells, and the cells were incubated for 48 h. p21 activity was measured by the luciferase reporter assay. (C) After the pRGC-luc reporter and the indicated genes were transfected in HeLa cells for 48 h, p53 activity was measured by the luciferase reporter assay. Data are presented as mean±SD (n=3). *p<0.05, **p<0.01, and ***p<0.001.
![]()
LC3 is conjugated to phosphatidylethanolamine (PE) through an enzymatic cascade involving Atg7, Atg3, and the Atg5-12-16 complex [28]. The unconjugated form of Atg8/LC3 (LC3-I) is found in the cytosol, while the conjugated form (LC3-II) targets the autophagosomal membrane [3328]. LC3-II remains in the membrane until it is degraded by lysosomes. The level of LC3-II detected by Western blotting is thus widely used as a marker of the autophagic process [28]. GFP-LC3-II is detected by immunoblotting with an antibody against GFP, and LC3-II protein level is correlated with the number of autophagosomes. We found that the LC3-II level was increased in ALFY- and TAK1-overexpressing cells, as compared with controls (Fig. 3A). Interestingly, ALFY was associated with an increase in LC3-II levels only in HEK293A and HeLa cells, but not in HCT116-/- cells, while TAK1-overexpressing cells showed higher LC3-II levels in all cell lines, including HEK 293A, HeLa, and HCT116-/-.
Fig. 3
TAK1 overexpression induces autophagy and p53 activation.
(A) HCT116 p53-/- and 293A cells were co-transfected with GFP-LC3 and the indicated constructs for 48 h. After transfection, cells were harvested and analyzed by immunoblotting. (B) HeLa cells were transfected with a mock plasmid or the indicated plasmids for 48 h and then harvested for immunoblot analysis. p53, p27, and p21 levels were evaluated using the appropriate antibodies. (C) p53 transcript levels in HeLa cells were analyzed by RT-PCR. GAPDH was used as an internal control. Data are presented as mean±SD (n=3). *p<0.05, **p<0.01, and ***p<0.001.
![]()
Autophagy/apoptosis induction can be either p53-dependent or -independent
Because p53 is known to be involved in cell cycle arrest, autophagy, and apoptosis, we investigated whether the 16 caspase-related genes affecting these processes are p53-dependent. Thus, we first attempted to unravel the molecular mechanisms underlying the regulation of p53 via the 16 caspase-related genes. In order to determine p53 dependency, we analyzed the effects of overexpression of these genes on a p53 luciferase reporter (pRGC-luc) harboring multiple p53 consensus sequences. Increasing ALFY, BIRC4, and TAK1 expression increased the activity of pRGC-luc in HeLa cells (Fig. 2C), suggesting that ALFY, BIRC4, and TAK1 increase the ability of p53 to transactivate the p53 reporter.
In another experiment, HeLa cells were transfected with each of the 16 caspase-related genes together with a p21-luciferase reporter (Fig. 2C). Increasing the levels of of 11 genes (ALFY, TSC22D3, CPSF1, RPS6KB1, UBE2D3, SEC61A2, N-PAC, BIRC4, TAK1, EEF1A1, and PDGFA) markedly induced the activity of the p21-luc reporter. These results suggest that p53-dependent (ALFY, TAK1, and BIRC4) and -independent mechanisms (SEC61A2, N-PAC, and WIPI1) may be responsible for the effects on p21 transactivation.
We further attempted to determine whether the 16 caspase-related genes can modulate endogenous p53 protein in HeLa cells by transfection with these genes. Interestingly, in HeLa cells, BIRC4 and TAK1 expression increased the level of endogenous p53 protein (Fig. 3B). Also, with the increased levels of p27 and p21 proteins, p53 targets were observed, corresponding with increases in p53 protein level.
To assess the mechanism underlying the increase in p53 modulated by BIRC4 and TAK1, quantitative RT-PCR was conducted on the p53 gene in order to determine whether BIRC4 and TAK1 induce p53 transcription. Transfection of BIRC4 and TAK1 in HeLa cells resulted in a significant increase in p53 mRNA levels (Fig. 3C). Also, transfection with BIRC4 and TAK1 increased p53 luciferase reporter activity in HeLa cells in a dose-dependent manner (Fig. 4A and B). Moreover, expression of TAK1 also increased the levels of endogenous p53 protein in HeLa cells in a dose-dependent manner (Fig. 4C). These results show that BIRC4 and TAK1 induce p53 mRNA transcription, thereby resulting in an increase in p53 protein expression.
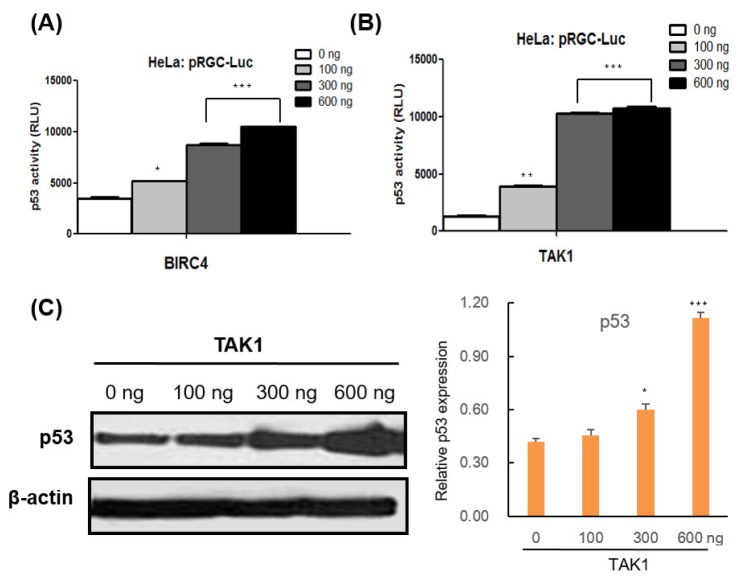
Fig. 4
The effects of BIRC4 and TAK1 on p53 activity.
(A) HeLa cells were transfected with the pRGC-Luc and BIRC4 plasmids. After 48 h, cell lysates were collected and assessed for luciferase activity. p53 activity increased in a BIRC4 dose-dependent manner. (B) HeLa cells were transfected with pRGC-Luc and TAK1 plasmids. After 48 h, cell lysates were collected and assessed for luciferase activity. p53 activity increased in a TAK1 dose-dependent manner. (C) HeLa cells were transfected with the indicated TAK1 plasmids. p53 protein expression level increased in a TAK1 dose-dependent manner. Data are presented as the mean±SD (n=3). *p<0.05, **p<0.01, and ***p<0.001..
![]()
DISCUSSION
Recently, autophagy, an evolutionarily conserved lysosomal degradation process, has been shown to play an important role in a variety of human diseases, such as cancer [29]. Notably, autophagy regulates several survival and cell death signaling pathways in cancer [3031]. To date, some key autophagic mediators, including p53, autophagy-related genes (ATGs), mTOR, and Beclin-1, are known to modulate autophagic activity in cancer initiation and progression [293031]. Because autophagy-modulating agents such as rapamycin and chloroquine have already been used clinically to treat cancer, it is conceivable that understanding and targeting autophagic pathways can provide new insights for the discovery of cancer therapeutics.
Apoptosis, the major form of controlled cell death, is involved in various diseases such as neurodegenerative diseases, autoimmune diseases, cardiovascular diseases, and cancer [29323334]. Therefore, investigations into the molecular mechanisms of apoptosis regulation have revealed a number of promising therapeutic targets and have been utilized in drug discovery and medical research. New therapeutic approaches to the regulation of apoptosis are currently being tested in preclinical and clinical trials.
SEC61A2 (Sec61 translocon Alpha 2) has a role in the insertion of secretory and membrane polypeptides into the endoplasmic reticulum [35]. It may also be required for the assembly of membrane and secretory proteins [35]. N-PAC, also known as GLYR1 (glyoxylate reductaste 1), and SEC61A2 have not been shown to induce apoptosis, but we reveal the novel finding that their expression increases apoptosis in HeLa cancer cells.
FALZ, also known as BPTF (Bromodomain PHD finger transcription factor), plays a role in transcriptional regulation, and high levels of FALZ have been detected in fetal brain tissue and in patients with neurodegenerative diseases [36]. Overexpression of FALZ in fibroblasts induces nuclear condensation and cleavage of caspase 3 to its active form, indicating induction of apoptosis [36]. Our data demonstrated a similar tendency by overexpression of FALZ in the HeLa cell line, but it did not affect autophagy.
ALFY, also known as WDFY3 (WD repeat and FYVE domain containing 3), functions as a master conductor for aggregate clearance by autophagy [37]. This protein is shuttled from the nuclear membrane to colocalize with aggregated proteins, where it complexes with other autophagic components to achieve macroautophagy-mediated clearance of these aggregated proteins. Reduced expression of ALFY and the formation of p62-positive polyubiquitinated protein aggregates promote cell death in RASFs under severe ER stress [37]. TAK1 (TGF-beta activated kinase 1), also known as MAP3K7 (Mitogen-Activated Protein Kinase Kinase Kinase 7), is a member of the serine/threonine protein kinase family [28]. This kinase mediates the signaling transduction induced by TGF beta and morphogenetic protein (BMP), and controls a variety of cellular functions, including apoptosis [38]. In a previous study, our group showed that TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1 [28]. Alfy and Tak1 are known to modulate autophagy as well as apoptosis; however, their involvement with the p53 pathway have not yet been reported. Our data demonstrate that ALFY and TAK1 induce apoptosis/autophagy in a p53-dependent manner, whereas SEC61A2 and N-PAC induce apoptosis/autophagy in a p53-independent manner. Overexpression of TAK1 increased p53 expression and accumulation of its target proteins as well as an increase in p53 mRNA levels.
The p53 pathway has been shown to mediate cellular stress responses, such as DNA repair, cell cycle arrest, senescence, autophagy, and apoptosis [3234]. These responses have been implicated in suppression of tumor formation and responses to many types of cancer therapy [34]. Apoptosis and autophagy usually function to eliminate damaged cells and damaged proteins, respectively [3334]. Dysfunction of these events is associated with oncogenesis and cancer progression. Therefore, our results can provide valuable information to better understand the apoptosis- and autophagy-related regulatory mechanisms of caspase-related genes and facilitate the exploitation of autophagy or apoptosis as targets for therapeutic intervention in cancer. Developing molecules that activate apoptosis and autophagy using validated targets will provide good candidates for anticancer therapeutics.
 XML Download
XML Download