

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Stress is a part of our existence. Stress makes us inefficient to manage our physical or psychological health. Stress can be caused by physical, psychological or social factors. Several clinical studies have shown that stress hyperglycemia was common in acute critically illnesses, even in patients without diabetes mellitus [1234]. Stress hyperglycemia occurs in 5~30% of patients with apoplexia, myocardial infarction, sepsis, trauma and other critical illness [2] and it correlates with poor outcome [5]. It has been well demonstrated that the elevation of stress state usually activates the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic nervous system (SNS). Increased activity of the SNS plays a role in the development of impaired glucose metabolism and lipid metabolism. blood pressure, heart rate, body temperature, plasma glucose, and adrenocorticotrophic hormone (ACTH) levels [67]. Activated sympathetic nervous system SNS causes a release of catecholamines (adrenalin and noradrenalin) from the adrenal medulla [8]. Catecholamines are very sensitive to stress ,and that adrenaline is a better reflection of stressor intensity than noradrenaline [9101112], and catecholamines are responsible for insulin resistance, on the receptor and post-receptor level or by inhibiting insulin release result in an increase of the blood glucose level by the stimulation of glycogenolysis and lipolysis [121314].
Stress raised the levels of hormones that stimulate the body to release stored glucose into the bloodstream this may further lead to hyperglycemia. In addition, HPA axis also plays a significant role in stress systems. The chief regulator of the HPA axis is a corticotrophin-releasing hormone (CRH). CRH is synthesized in the hypothalamus [15], and CRH stimulates the secretion of an ACTH and subsequently, promotes glucocorticoid secretion from the adrenal cortex [16]. It has been well demonstrated that glucocorticoid, in general, causes the hyperglycemia by activating a gluconeogenesis pathway [17], whereas sympathetic nervous system activation causes the blood glucose level by activating glycogenolysis [14]. The end effect of this is that more energy will be available to the body in the form of glucose to fight against stress. Excessive exposure to stress hormones such as glucocorticoid can damage the brain [18]. Numerous studies have also suggested that hyperglycemia may result in neuronal cell death and glial damage. Hyperglycemia during ischemia may cause anaerobic glycolysis, which may lead to intracellular acidosis to, in turn, cause an increase in cerebral lactate concentration, finally result in neuronal and glial damage [192021]. Hyperglycemia and hypoglycemia have been considered as keys for the survival or death of neuronal cells in the brain.
Some studies have shown that PTX treatment resulted in an inhibition of both insulin binding and glucose transport [2223], this may further decrease glucose level. We have also recently reported In our previous studies, we have reported that i.t. administration with PTX causes hypoglycemia in a comprehensive manner and induced neuroprotection against the KA-induced hippocampal CA3 neuronal death by lowering the blood glucose level [2425]. Although PTX is involved in the regulation of the blood glucose level, However, the exact role of PTX-sensitive G-proteins in stress-induced hyperglycemia has not been well characterized yet. Certain stress models such as the restrain-, cold water swimming stress-, an electric foot shock physical- and witness emotional-stress animal models are often adopted for researching stress-related mechanisms. Thus, in the present study, the effect of pretreatment with PTX administered supraspinally or spinally on the blood glucose level induced by several types of stresses such as restraint-, cold-water swimming-, electric foot-shock physical-, and witness emotional-stress was examined.
METHODS
These experiments were approved by the Hallym University Animal Care and Use Committee (Registration Number: Hallym 2009-05-01). All procedures were conducted in accordance with the 'Guide for Care and Use of Laboratory Animals' published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Stress.
Experimental animals
Male ICR mice (MJ Co., Seoul, Korea) weighing 20~25 g were used for all the experiments. Animals were housed 5 per cage in a room maintained at 22±0.5℃ with an alternating 12 h light-dark cycle. Food and water were available ad libitum. The animals were allowed to adapt to the laboratory for at least 2 h before testing and were only used once. Experiments were performed during the light phase of the cycle (10:00~17:00).
Intracerebroventricular and intrathecal injections
The i.c.v. injections were performed following the procedure established by Laursen and Belknap [26]. Each mouse was grasped firmly without anesthesia by the loose skin behind the head. The skin was pulled taut. Each mouse was injected with drug manually at bregma with a 50 µl Hamilton microsyringe fitted with a 26-gauge needle that was inserted to a depth of 2.4 mm. The i.c.v. injection volumes were 5 µl, and the injection sites were verified by injecting a similar volume of 1% methylene blue solution and determining the distribution of the injected dye in the ventricular space. The experiments were performed only when the success rate of i.c.v injection was over 95%. I.t. administration was performed in conscious mice following the method of Hylden and Wilcox [27] using a 30-gauge needle connected to a 25 µl Hamilton syringe with polyethylene tubing. The i.t. injection volume was 5 µl, and the injection site was verified by injecting a similar volume of 1% methylene blue solution and determining the distribution of the injected dye in the spinal cord. All injections were performed manually at the level of lumbar vertebra (L4~L5). The dye injected i.t. was distributed both rostrally and caudally but with short distance (about 0.5 cm) and no dye was found in the brain. The success rate for the injections was consistently found to be over 95% before the experiments were done.
Electrical foot shock-witness model
A two-compartment box, as described by Van den Berg et al. was used [28]. An electrical foot shock was applied to the physical stress animal group in one chamber, whereas the witness animal group in another chamber did not receive any electrical foot shock and was regarded as an emotional stress model. The perforated plexiglass between two compartments enabled mice to do visual, auditory, and olfactory communication. The mice were exposed to one of two conditions: physical stress, emotional stress. Unpredictable foot shocks (0.25 mA, 1s) to the physical stress group were delivered at random for 10 min total 10 times. The emotional stress group was present in the adjacent compartment during foot shock treatment. The physical stress group was present in the same compartment as the physical stress group without receiving any foot shock. The emotional stress group was also placed in the same compartment as the emotional stress group without receiving any witness stress. The stress was applied unpredictably at the rate of 10 stimuli/10 min/day. Shocks were delivered through the metal grating and generated by a shock generator and scrambler. The two compartment box was cleaned with water and soap after each pair of mice.
Immobilization stress model
The mice were subjected to restraint stress as described in a previous study [29]. In brief, restraint was carried out by placing the mouse in a 50 ml Corning tube, and adjusting it with an iron nail on the outside, which crossed in the caudal part of the animal. Adequate ventilation was provided using holes at the sides of the tubes. The mice were stressed by restraint for 30min. In the stress model, there was a single exposure.
Cold-water swimming stress model
The mice were forced to swim in cold (4℃) water for 3 min. The mice were allowed to swim in a container 15 cm in diameter and 20 cm tall with water filled to a depth of 11 cm. After the swimming, the mice were gently dried by patting the body with a paper towel.
Measurement of blood glucose level
The blood glucose level as measured at 30, 60, 90 and 120 min after stress by tail-vein blood collection in mice. The blood sampling was progressed at short time, and it needed the blood of 1 µl. The blood glucose level was measured using an Accu-Chek Performa blood glucose monitoring system (Mannheim, Baden-Württemberg, Germany).
Measurement of blood glucose level
The blood glucose level was measured at 30, 60 and 120 min after administration (n=8~10). The blood was collected shortly as soon as possible with a minimum volume (1 µl) from the tail-vein. The glucose level was measured using Accu-Chek Performa blood glucose monitoring system (glucometer) (Mannheim, Baden-Württemberg, Germany).
Insulin ELISA assay
In Mouse Insulin ELISA, biotin-conjugated anti-insulin, and standard or sample are incubated in monoclonal anti-insulin-coated wells to capture insulin bound with biotin conjugated anti-insulin. After 2 h incubation and washing, HRP (horseradish peroxidase) conjugated streptavidin is added, and incubated for 30 min. After washing, HRP conjugated streptavidin remaining in wells are reacted with a substrate chromogen reagent (TMB) for 20 min, and the reaction is stopped by the addition of acidic solution, and the absorbance of the yellow product is measured spectrophotometrically at 450 nm. The absorbance is proportional to insulin concentration. The standard curve is prepared by plotting absorbance against standard insulin concentrations. Insulin concentrations in unknown samples are determined using this standard curve.
Corticosterone assay and blood sampling
The plasma corticosterone level was determined by the f luorometric determination method [30]. Four hundred microliters of blood were collected by puncturing the retro-orbital venous plexus. Plasma was separated by centrifugation and stored at 80℃ until assayed.
Drugs
PTX was purchased from Research Biomedicals Inc. (Natick, MA, USA). PTX was dissolved in saline, and it was prepared just before use.
Statistical analysis
Statistical analysis was carried out by Anova (Bonferroni test) for multiple comparisons and student t-test by using GraphPad Prism Version 4.0 for Windows (GraphPad Software, San Diego, CA, USA). p-values less than 0.05 were considered to indicate statistical significance. All values were expressed as the mean±S.E.M. In our study, we established the mean blood glucose value of the control group through many experiments under matching conditions. Selected mice of established blood glucose level were then used in replication experiments.
RESULTS
Effect of PTX on the blood glucose, corticosterone, and insulin levels induced by an electric foot-shock physical stress and witness stress
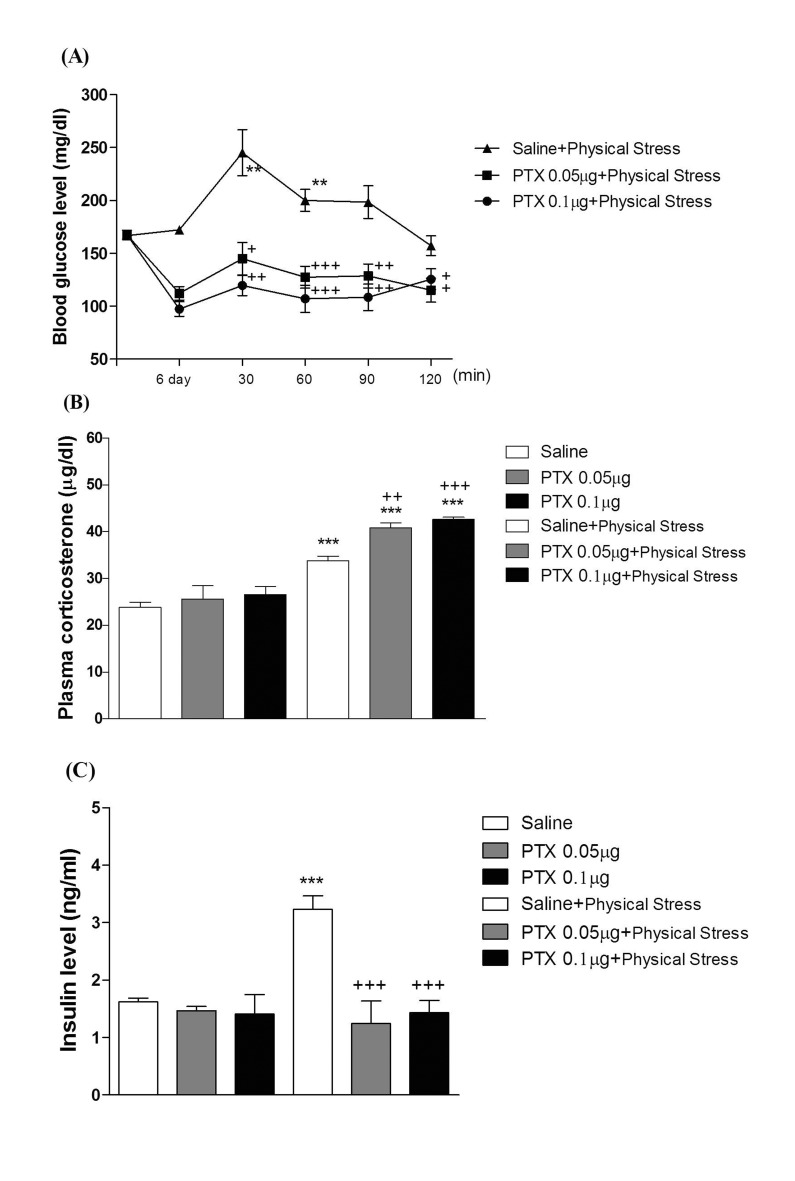
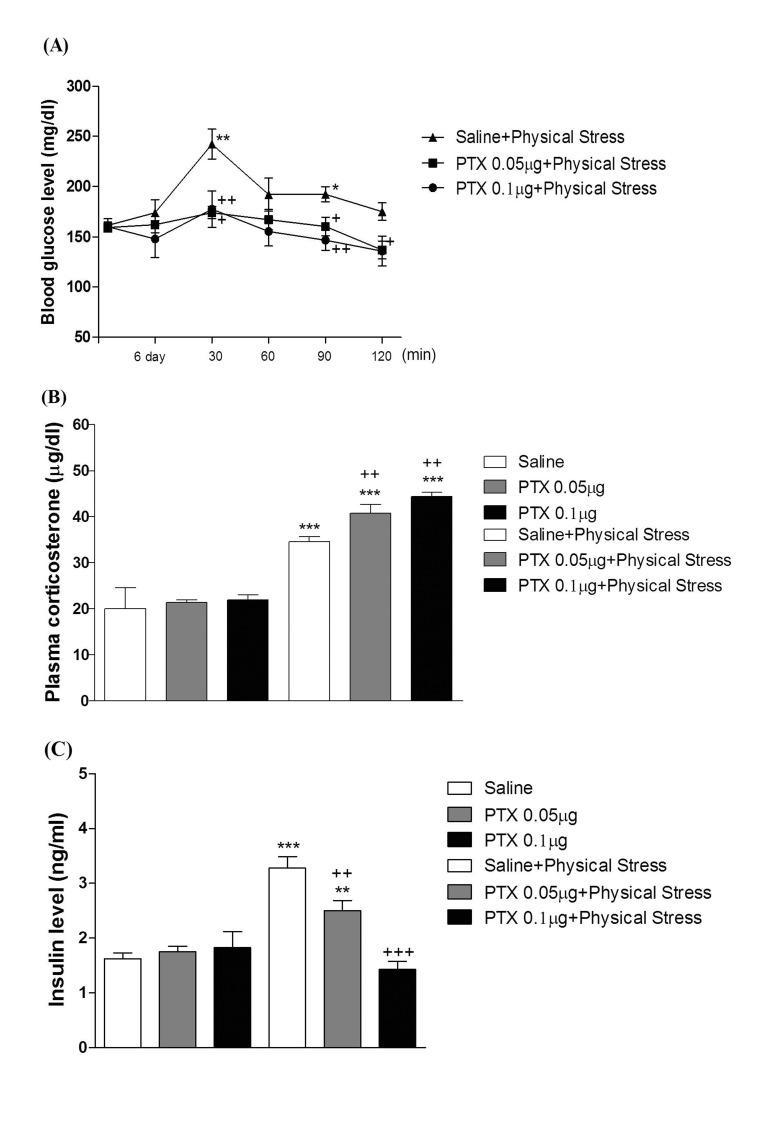
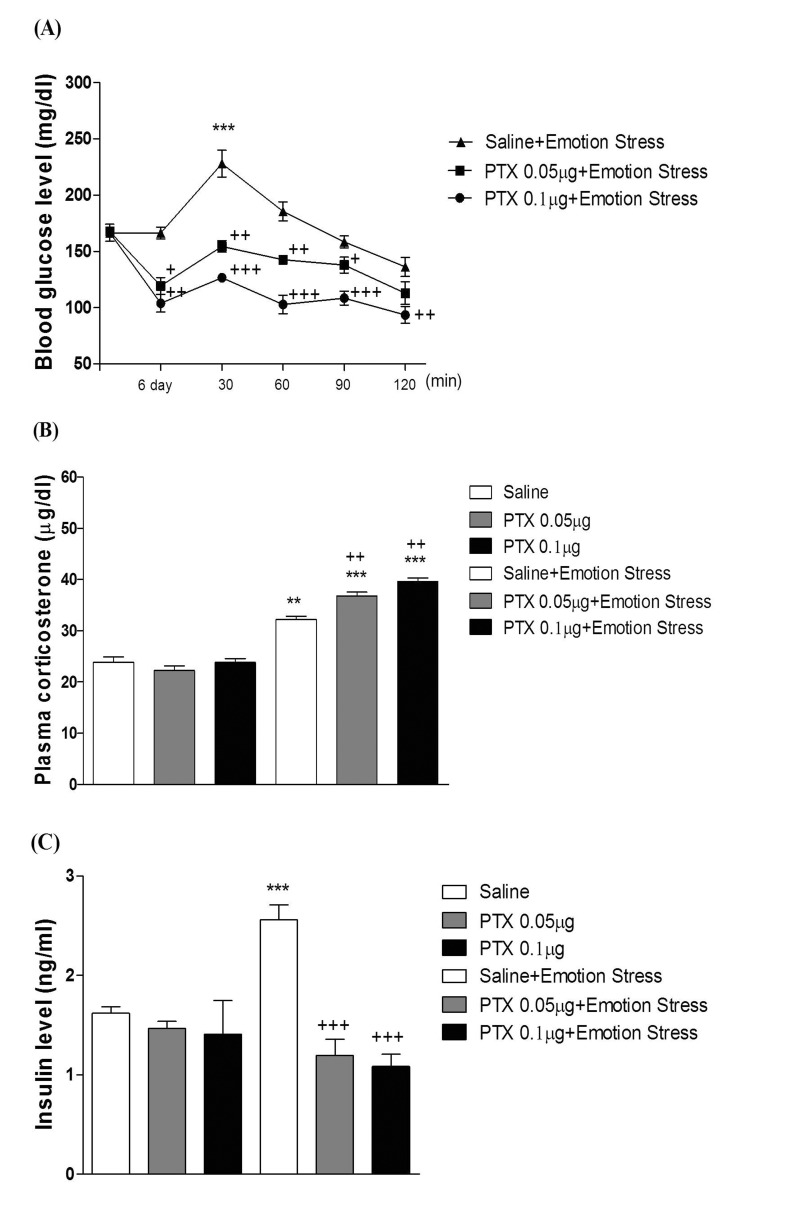
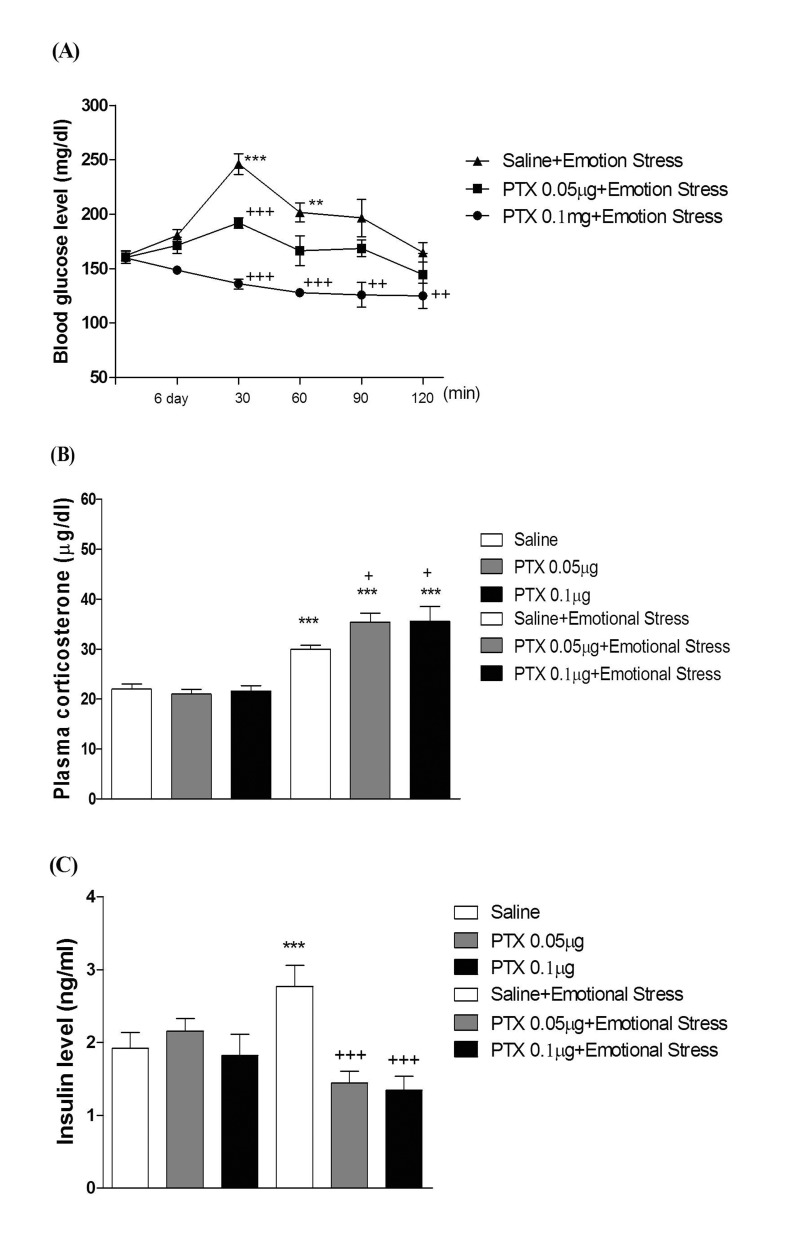
Mice were exposed to physical stress after the pretreatment of PTX (0.05 or 0.1 µg) i.c.v. or i.t. once for 6 days. Blood glucose level was increased by electric foot-shock stress stimulation. The blood glucose level reached the maximum level at 30 min after and returned to the basal level at 120 min after stress stimulation. Administration of PTX i.c.v. or i.t. significantly inhibited the physical stress-induced blood glucose level in a dose-dependent manner (Fig. 1A and 2A respectively). The high dose of PTX (0.1 µg/kg) almost abolished the blood glucose level induced by physical stress stimulation. In addition, the blood glucose level was also elevated in emotional stress group. The blood glucose level reached the maximum level at 30 min and decreased to a basal level at 120 min after stress stimulation. I.c.v. or i.t pretreatment with PTX attenuated the elevation of emotional stress-induced blood glucose level in a dose-dependent manner (Fig. 3A and 4A respectively). Moreover, the plasma corticosterone and insulin levels were up-regulated in both physical and emotional stress group. Whereas i.c.v. or i.t. pretreatment with PTX caused inhibition of the blood insulin level in both physical (Fig. 1C and 2C), and emotional stress (Fig. 3C and 4C) groups in a dose-dependent manner. However, i.c.v. or i.t. pretreatment with PTX caused a further elevation of the blood corticosterone level in both physical (Fig. 1B and 2B) and emotional stress groups in a dose-dependent manner (Fig. 3B and 4B).
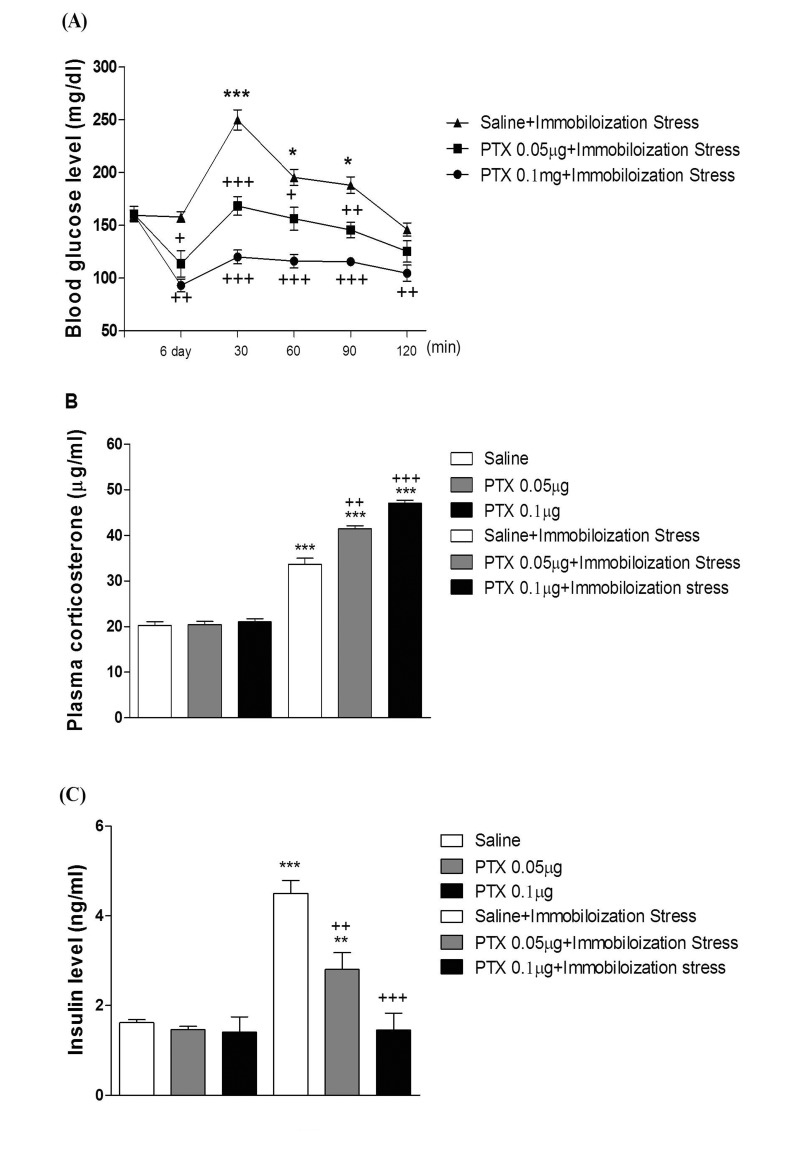
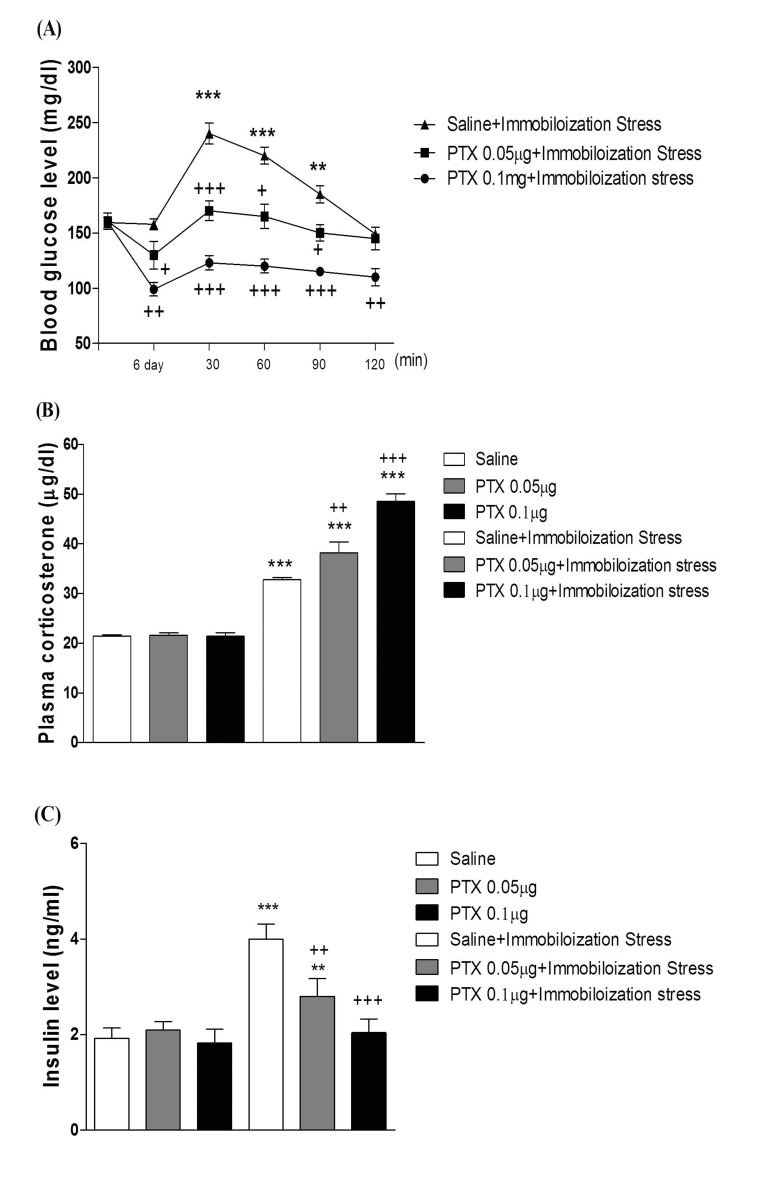
Effect of PTX on the blood glucose, corticosterone, and insulin levels induced by immobilization stress
Mice were pretreated i.c.v. or i.t. once for 6 days with PTX (0.05 or 0.1 µg). Then mice were enforced into immobilization stress for 30 min and returned to the cage. The blood glucose level was measured at 30, 60 and 120 min after immobilization stress initiated. As shown in Fig. 5A and 6A, PTX pretreated i.c.v. or i.t. attenuated the elevation of the blood glucose level induced by immobilization stress in a dose-dependent manner. In addition, the blood corticosterone and insulin levels were measured at 30 after immobilization stress initiated. Both plasma corticosterone and insulin levels were up-regulated in immobilization stress (Fig. 5B, 5C, 6B, and 6C). The plasma insulin level was decreased by PTX pretreated i.c.v. or i.t. as shown in Fig. 5C and 6C, whereas the i.c.v. or i.t. pretreatment with PTX caused an up-regulation of the blood corticosterone level (Fig. 5B and 6B).
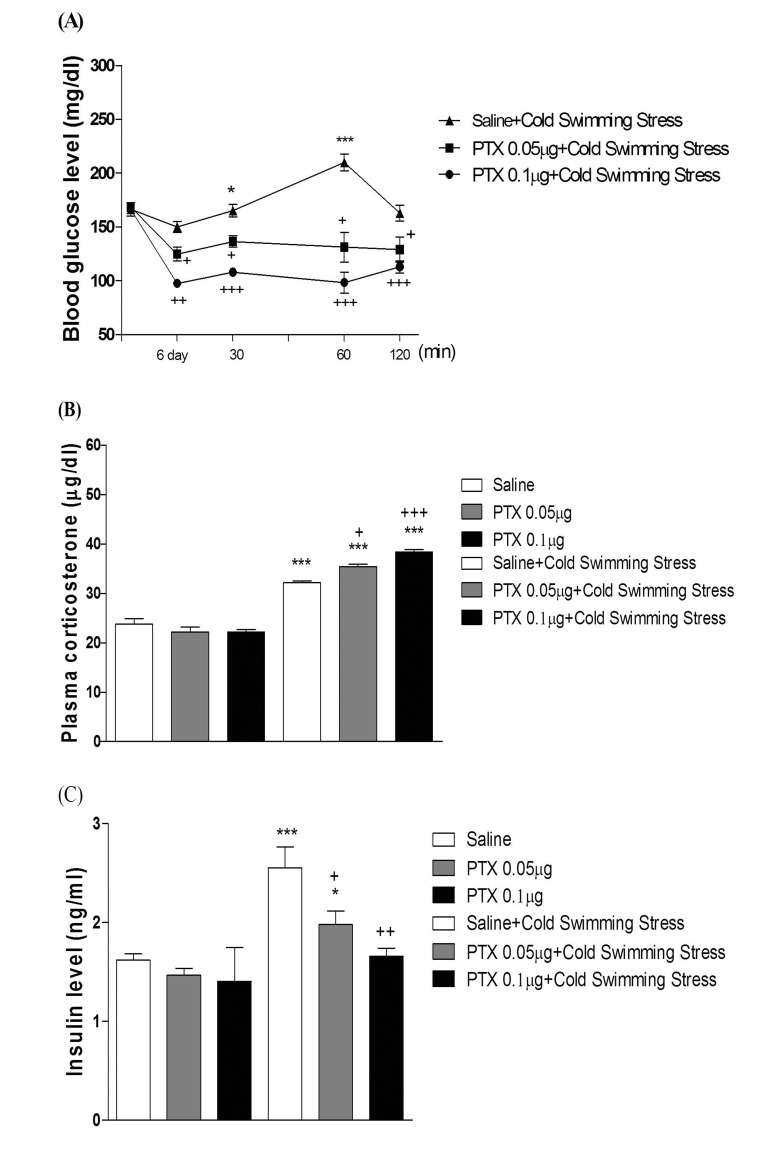
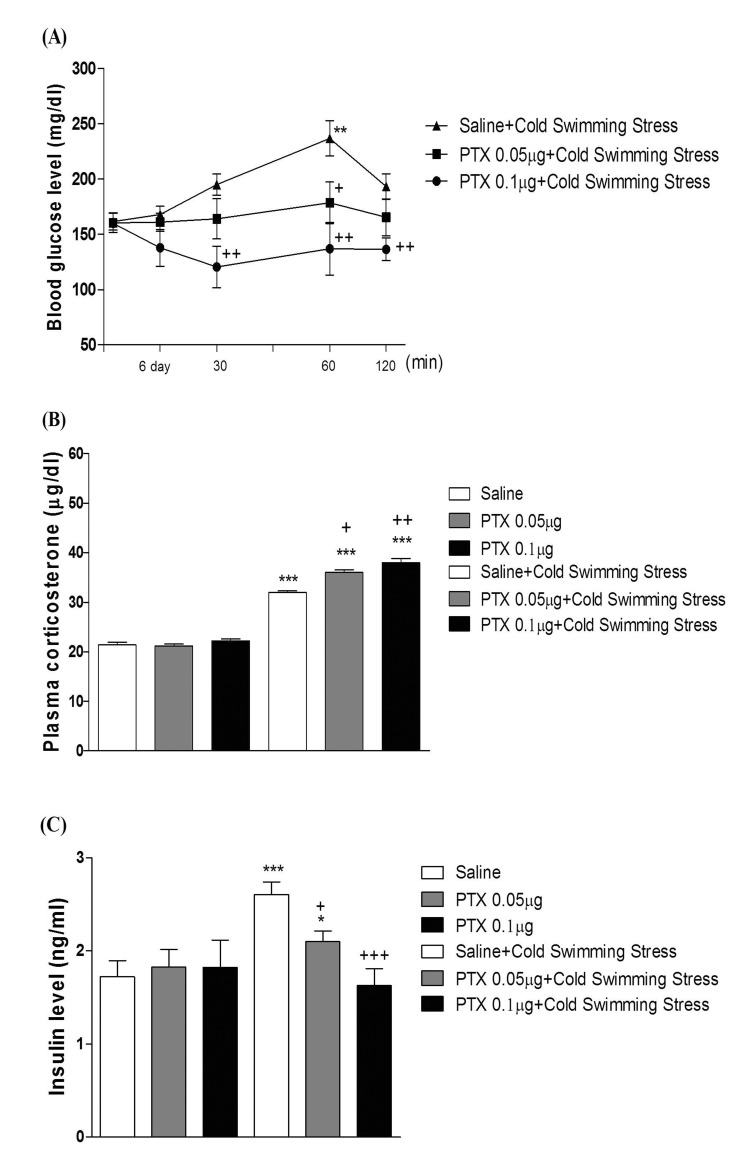
Effect of PTX on the blood glucose, corticosterone, and insulin levels induced by cold-water swimming stress
Five min after the swimming, mice were subjected to measurement of blood glucose level. As shown in Fig. 7A and 8A, swimming at 4℃ caused a slight elevation of the blood glucose level. The blood glucose level was gradually increased up to 60 min and returned to the control level at 120 min after cold-water swimming stress initiation. As shown in Fig. 7A and 8A, PTX pretreated i.c.v. or i.t. attenuated the elevation of the blood glucose level induced by cold-water swimming stress in a dose-dependent manner. In addition, the blood corticosterone and insulin levels were measured at 60 min after cold-water swimming stress initiated. Both plasma corticosterone and insulin levels were up-regulated in cold-water swimming stress (Fig. 7B, 7C, 8B, and 8C). The plasma insulin level was decreased by PTX pretreated i.c.v. or i.t. (Fig. 7C and 8C), whereas i.c.v. or i.t. pretreatment with PTX caused an up-regulation of the blood corticosterone level (Fig. 7B and 8B).
DISCUSSION
In previous studies, we and others have observed that blood glucose level is increased in various types of animal stress models [31323334]. It has generally been known that the hyperglycemic effect is induced by both physical and emotional stress. For example, it has been reported that blood glucose level is significantly increased in foot shock stress model [353637]. In addition, recently we found that the alteration of blood glucose level in emotional stress is evidenced, suggesting that the blood glucose level is regulated by both physical and psychological stresses [34]. Furthermore, in the present study, we found that supraspinal or spinal pretreatment with PTX effectively causes a reduction of the increased blood glucose level induced by physical, emotional, immobilization or cold-water swimming stress, suggesting that PTX-sensitive G proteins located at supraspinal and spinal levels appear to be involved in stress-induced hyperglycemia.
The roles of adrenergic receptors in the regulation of modulation of stress processing have been demonstrated in several previous studies. For example, as stress-induced hyperglycemia appears to be strongly related to adrenaline release, the adrenaline release is a sensitive marker of adaptation to repeated stress [383940]. In a previous study, we demonstrated that the i.p. injection of α-adrenergic receptor antagonists such as phentolamine and yohimbine significantly inhibited the blood glucose level induced by both physical and emotional stress [34]. However, the present study was designed to find out the role of supraspinally and spinally located PTX sensitive G-proteins in blood glucose regulation under various types of stress conditions. We and others have previously demonstrated that PTX administered systemically or centrally exerts a hypoglycemic effect [254142]. In addition, i.t. pretreatment with PTX also dose-dependently attenuated the blood glucose level induced by baclofen [42]. Furthermore, it has been reported that hyperglycemia induced by clonidine, an α2-adrenergic receptor agonist, is blocked by PTX pretreated spinally, suggesting that the activation of PTX-sensitive inhibitory G-proteins by clonidine may cause hyperglycemia [43]. In the present study, the blockade of hyperglycemia induced by physical and emotional stresses by PTX pretreated supraspinally or spinally suggests that the activation of the PTX-sensitive G-proteins may be one of the mechanisms involved in stress-induced hyperglycemia.
Recent lines of study directly raised the possibility that the hyperglycemia induced by the restraint stress for 6 h is suppressed by pre-administration with RU-486 [44]. In the current study, we examined the possible inhibitory action of PTX pretreated supraspinally or spinally against the elevation of the blood glucose level induced by immobilization stress. We found that supraspinal or spinal pretreatment with PTX exerted an inhibitory action on the increased blood glucose level induced by immobilization stress. In addition, immobilization stress-induced plasma insulin level was decreased by PTX pretreated supraspinally or spinally, suggesting that the reduction of the blood glucose level by i.c.v. or i.t. pretreatment with PTX appears to be due to a reduction of stress-induced insulin level. However, we found in the present study that PTX pretreated supraspinally or spinally caused a further elevation of the blood corticosterone level induced by immobilization stress, indicating that the up-regulation of the plasma corticosterone by PTX in immobilization stress model appears to be due to a blood glucose homeostatic mechanism.
Moreover, the action of PTX pretreated supraspinally or spinally against the elevation of the blood glucose level induced by cold-water swimming stress was also assessed. We found in the present study that the cold-water swimming causes a gradual elevation of the blood glucose level right after the cold-water swimming. The blood glucose level began to increase gradually up to 60 min and returned to the control level 120 min after the cold-water swimming. Cold-water swimming stress also causes the elevation of the plasma corticosterone and insulin levels. The supraspinal or spinal pretreatment with PTX causes a reduction of the plasma insulin level induced by cold-water swimming stress, suggesting that PTX pretreated supraspinally or spinally attenuates the blood glucose level induced by cold-water swimming stress by reducing the plasma insulin level. However, the supraspinal or spinal pretreatment with PTX causes an up-regulation of the plasma corticosterone level induced by cold-water swimming stress, indicating that the plasma corticosterone level up-regulation by supraspinal or spinal pretreatment with PTX appears to be due to a blood glucose homeostatic mechanism.
In conclusion, the present study clearly shows that the foot shock stimulation-induced physical and emotional, immobilization and cold-water swimming stress increases the blood glucose level. Hyperglycemia produced by various stress appears to be mediated by activation of supraspinally or spinally located PTX-sensitive G proteins. The reduction of blood glucose level by PTX seems to be due to the reduction of plasma insulin level. And up-regulation plasma corticosterone level by PTX in stress models may be due to a blood glucose homeostatic mechanism.
 XML Download
XML Download