

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dexmedetomidine (DEX) is a methylol derivative with high affinity for the α2-adrenoceptor that is used widely in the perioperative period for its sedative, analgesic, sympatholytic and anxiolytic actions [1].
Cardiovascular side effects of DEX have been reported, including hypotension, hypertension and decreased heart rate [2]. In addition to its sympatholytic effects mediated by central presynaptic α2-adrenoceptors and on decreasing arterial blood pressure and heart rate, DEX has effects on peripheral blood vessels [345]. Activation of postsynaptic α2-adrenoceptors would induce both vasoconstriction (through the activation of α2-adrenoceptors on vascular smooth muscle cells) and vasodilatation (through the activation of α2-adrenoceptors on endothelial cells) had been proved by previous studies [678]. Resulting from the interaction between the actions of central presynaptic α2-adrenoceptors and peripheral postsynaptic α2-adrenoceptors, the vascular effects of DEX are complex as a rule [9]. In contrast to α1-adrenergic receptors, α2-adrenergic receptors are expressed at high density in only a number of tissues such as the human femoral vein and artery [10]. Experimental findings show that the localization of adrenergic receptor subtypes can differ among species and vessel segments, resulting in variation in the responses to the activation of these receptors. Studies in vivo showed that high doses of DEX induced pulmonary hypertension [11]. however, the actions of DEX on human pulmonary resistance arteries in vitro have not been reported to date.
We studied the effects of DEX on human pulmonary arteries in preparation and found that DEX had no effect on endothelium-intact pulmonary arteries, whereas high concentrations of DEX elicited contractions in endothelium-denuded pulmonary arteries. A conceivable reason for that is α2-adrenoceptors in pulmonary arteries would produce a balance between direct smooth muscle-dependent vasoconstriction and endothelium-dependent vasodilatation.
The current study was designed to test the hypothesis that DEX inhibits vascular tension in human pulmonary arteries through the endothelial nitric oxide (NO) synthase dependent production of NO.
METHODS
Patients
This study was performed after obtaining approval from the Research Ethics Committee of Guangdong General Hospital (NO.GDREC2014023H(R1)). Peripheral human lung tissue was obtained from 62 patients (35 men and 27 women; age range 24~67 years; including 23.81% smokers) who underwent pulmonary lobectomy for lung carcinoma. The patients showed no clinical evidence of hypertension, pulmonary hypertension or diabetes, and none of them had received steroids or nonsteroidal anti-inflammatory drugs before surgery.
Preparation of pulmonary arterial rings
During transport to the laboratory, lung tissues were placed in ice cold Krebs solution with the following composition (in mM): NaCl, 118; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; KH2PO4, 1.2; NaHCO3, 25; and D-glucose, 11.1 (pH 7.4). Fat and connective tissues were carefully removed under a binocular microscope, and a vascular ring (length, 2 mm; diameter, 1~1.5 mm) was prepared from each artery for tension recording. Two stainless wires (each 40 µm in diameter) were introduced through the lumen of the vascular ring and each wire was fixed to the jaws of a myograph. The organ chamber was filled with 5 ml Krebs solution, which was gassed with 95% O2 and 5% CO2 at 37℃. Changes in arterial tension were recorded isometrically by a Multi Myograph System (Danish Myo Technology, Aarhus, Denmark). Each ring was stretched gradually to a resting tension of 1.8~2 mN during a 90 min equilibration period to obtain optimal tension. The Krebs solution was replaced in the chamber every 20 min and tension was readjusted to 1.8~2 mN. In some rings, we Insetted a small forceps into the lumen cautiously and rolled the ring backward and forward to denude the endothelium.
There were one to four rings per patient, and only one ring in each group came from the same patient. Each preparation was used in only one experimental protocol.
Vascular reactivity test
After the equilibration period, 60 mM of KCl was added to the baths and the contractile force was recorded. The vascular rings were washed four times with Krebs solution at intervals of 5 min to restore the basal tension. Changes in vascular tone amplitude of <3 mN were considered indicative of poor contractibility and the arterial rings were discarded.
Endothelial integrity test
Endothelial integrity in the pulmonary arterial rings was confirmed by at least 80% relaxation in response to acetylcholine (ACh, 10–6 mol/L) at the plateau of serotonin (10–5 mmol/L). Successful endothelial removal was confirmed by a lack of vasorelaxant response to ACh. To exclude any residual effect of Ach, the following experimental protocols were performed 60 min after the Ach trials. Only preparations that responded adequately to the serotonin challenge (>3 mN) were used for further examinations. There were one to four rings per patient, and only one ring in each group came from the same patient. Each preparation was used in only one experimental protocol.
Effect of DEX on pulmonary arterial rings
DEX (10–10~10–6 mol/L) was added to intact and endothelium-denuded rings cumulatively and a cumulative concentration-response curve (CRC) for DEX was obtained.
Effect of serotonin on pulmonary arterial rings
Serotonin (10–8~10–5 mol/L) was added to intact and endothelium-denuded rings cumulatively, and the tension was expressed as a percentage of the initially contraction of serotonin (10–5 mol/L). A cumulative CRC for serotonin was obtained. The first CRC to serotonin was not altered by the previous challenge with serotonin (10–5 mol/L), whereas consecutive CRCs in the same preparation showed a depression compared to the initial curve [12]. Therefore, a single CRC to an agonist was generated in each preparation either in the absence or presence of a single concentration of an antagonist.
Effect of DEX on pulmonary vasoconstriction induced by serotonin
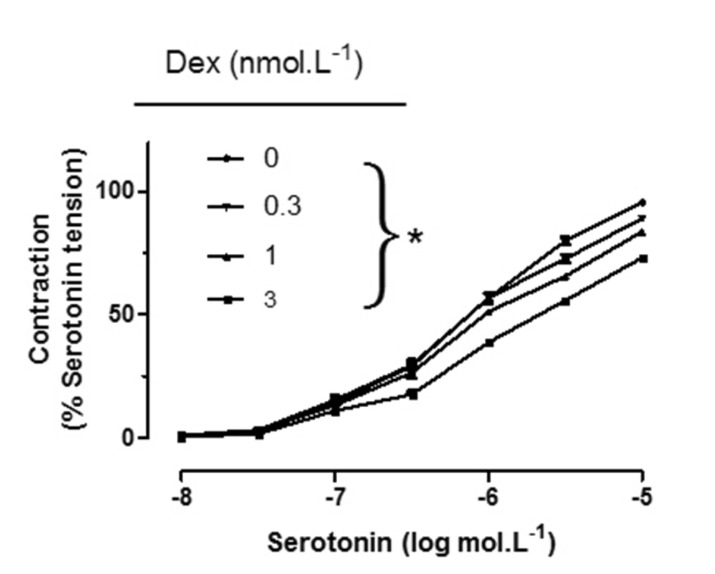
Rings were incubated for 30 min with DEX at 3×10–10, 10–9, and 3×10–9 mol/L, and the changes in tension were measured. To determine the effect of DEX on contractility, a CRC to serotonin was obtained while maintaining the doses of DEX at 3×10–10, 10–9, and 3×10–9 mol/L.
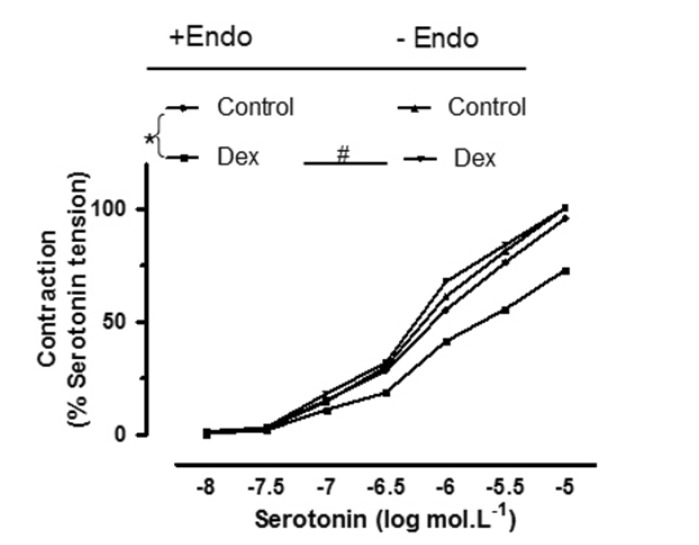
Effect of different treatments and endothelium on the inhibition of pulmonary vasoconstriction by DEX
Cumulative concentrations of serotonin were added to intact rings incubated with DEX (3×10–9 mol/L) in the presence or absence of L-NAME (10–4 mol/L), yohimbine (5×10–8 mol/L), and indomethacin (5×10–6 mo/L). In endothelium-denuded rings, cumulative concentrations of serotonin were added without any treatments after incubation with DEX (3×10–9 mol/L).
Statistical analysis
Data were expressed as the mean±SEM of n experiments and analyzed with Student's t-test or two-way ANOVA followed by Bonferroni posthoc tests when more than two groups were compared. The contraction response was expressed as a percentage of the initial contraction of serotonin (10–5 mol/L). Emax represented the maximal response percentage. pD2 represented the negative logarithm of the vasoconstrictor concentration required to produce half of the maximal contraction. pD2 and Emax were determined by non-linear regression curve fitting (Graphpad Prism software, version 5.0). p<0.05 was considered statistically significant.
RESULTS
Effect of DEX on pulmonary arterial resting tension
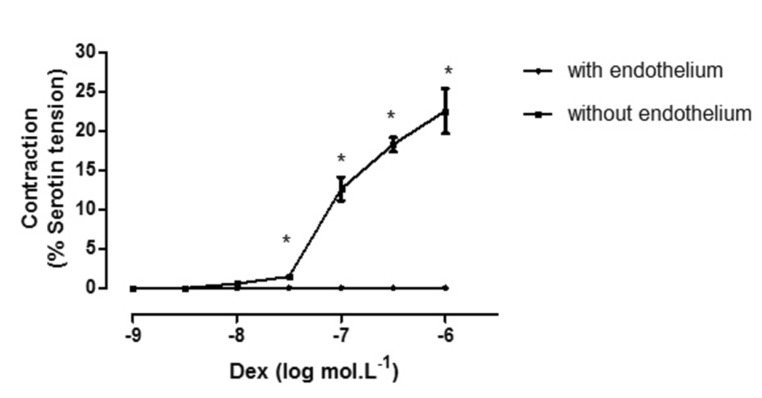
DEX (10–10~10–6 mol/L) had no direct effect on the resting tension of endothelium-intact pulmonary arteries. However, DEX (10–8~10–6 mol/L) caused concentration-dependent contractions in endothelium-denuded human pulmonary arteries (Fig. 1).
Effect of serotonin on pulmonary arterial resting tension
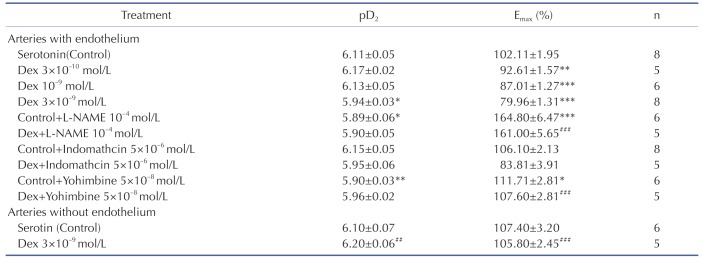
Serotonin produced a concentration-dependent contraction either in endothelium-intact pulmonary arteries (pD2: 6.11±0.05, Emax: 102.10±1.96%) or endothelium-denuded pulmonary arteries (pD2: 6.10±0.07, Emax: 107.40±3.20%) (Table 1).
The inhibitory effect of DEX on contraction is mediated by NO
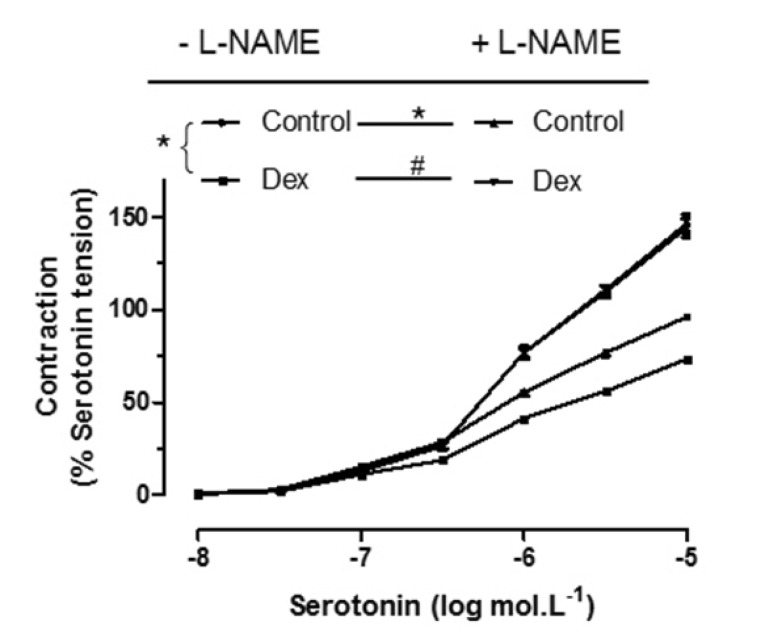
L-NAME is an endothelial nitric oxide synthase inhibitor. Incubating of L-NAME (10–4 mol/L) could abolish the inhibitory effect of DEX (3×10–9 mol/L) on serotonin-induced contraction (Fig. 4), which indicated that the inhibitory effect of DEX was mediated by nitric oxide synthase.
The inhibitory effect of DEX on contraction is mediated by α2-adrenoceptor activation
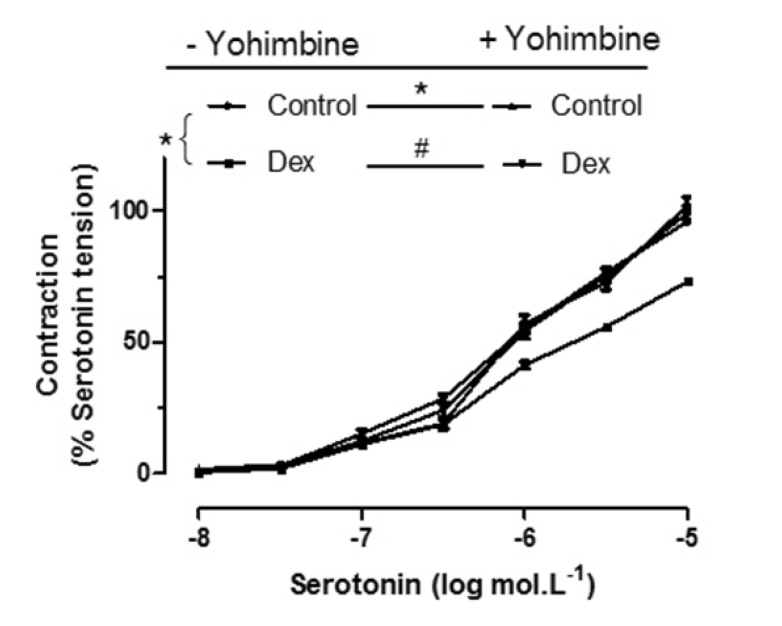
Yohimbine is an α2-adrenoceptor antagonist. The DEX (3×10–9 mol/L) inhibitory response is significantly abolished by Yohimbine (5×10–8 mol/L). The serotonin-induced contraction curve is more great when incubated with DEX and yohimbine (Fig. 5).
The inhibitory effect of DEX on contraction is unrelated to the cyclooxygenase pathway
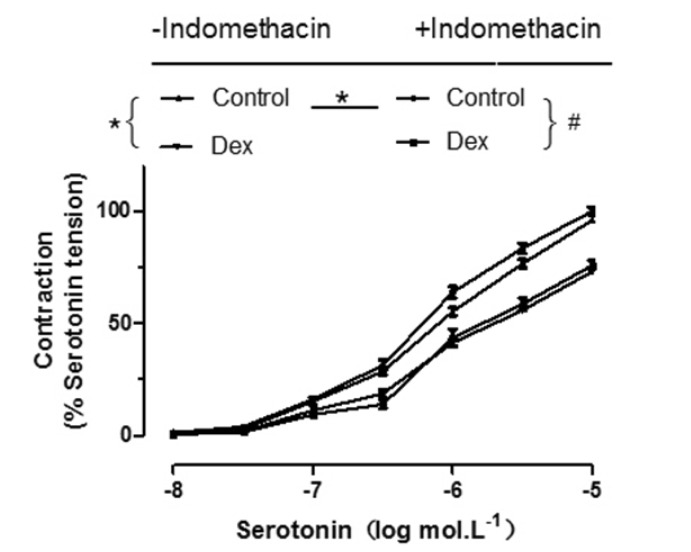
The inhibitory effect of DEX (3×10–9 mol/L) on serotonin-induced contraction is not significantly changed in the absence or presence of indomethacin which is a cyclooxygenase inhibitor (Fig. 6).
DISCUSSION
The main finding of the present study was that DEX inhibited vascular tension in human pulmonary arteries, and this effect was abolished by L-NAME, endothelial denudation or yohimbine, suggesting that the DEX-induced inhibition of vasoconstriction in human pulmonary arteries is mediated by the production of nitric oxide (NO) induced by the activation of endothelial α2-adrenoceptors and nitric oxide synthase.
DEX induces vasoconstriction in humans and animals [131415161718] by increasing in arterial pressure and peripheral vascular resistance. The constrictive effects of DEX have been observed in dog [13], pig [14] and goat coronary arteries [15], rat pial arterioles [16], human gastroepiploic artery [17], internal mammary artery [18] and brachial arteries [19]. As these studies have shown, stimulation of α2-adrenoceptors present in vascular smooth muscle would cause vasoconstriction, which can also be incurred by the stimulation of α1-adrenoceptors by high concentrations of DEX.
Nevertheless, due to the variableness in the sensitivity to DEX, the extent of the retraction caused by this agent differs greatly among the different vessels [13]. For instance, Synapair et al. [20]. Showed that endothelial NOS inhibition is required for the indication of DEX-induced vasoconstriction of arteries, yet venous constriction does not require such treatment [21]. Yildiz [18] and Won-Sung Kim et al. [22] indicated that clinical doses of DEX could not induce vasoconstriction in the human internal mammary artery and gastroepiploic artery. These findings are contrary to those of previous clinical trials.
Activation of NOS by α2-adrenoceptor agonists has been shown in several animal models and in humans [6713202324]. Kim et al. [7] showed that severe vasoconstriction caused by high doses of DEX was prevented by DEX-related NO production. In another study, DEX induced NO production in human umbilical vein endothelial cells [24]. Coughlan et al. [13] showed that NOS inhibition enhances DEX-induced constriction of coronary arteries, although it has no effect on cerebral arteries. In a clinical study conducted in healthy volunteers, therapeutic doses of DEX produced vasodilation mediated by eNOS in contrast to its known vasoconstrictor effects [20]. Wong et al. [6] reported that DEX induced relaxation at low concentrations (10–12 mol/L~3×10–8 mol/L) followed by contraction at higher concentrations (>3×10–8 mol/L) in the rat mesenteric artery. The relaxation in response to DEX was dependent on NO and the endothelium, and it was enhanced in the presence of indomethacin. These authors also showed that the vascular effects of DEX depend on the type of blood vessel because DEX did not induce endothelium-dependent relaxation in the rat aorta. Blood vessels have a wide variety of postsynaptic α1- and α2- adrenoceptors. The correlation between arterial diameter and the presence of α2-adrenoceptors has proved to be inverse as it appears, because large arteries have both α1- and α2- adrenoceptors, while α2-adrenoceptors are more eminent in small arteries and veins [25]. Given all that, these findings reveal that the effect of DEX on systemic vascular resistance and blood pressure rest upon dose, context and patient, and the balance between direct smooth muscle-dependent vasoconstriction and endothelium-dependent vasodilatation caused by DEX may differ among different organs.
Our group has done a lot of work on isolated pulmonary arteries [26], the present study is the first to investigate the effect of DEX on human pulmonary arteries. We showed that DEX (10–8~10–6 mol/L) caused concentration-dependent contractions in endothelium-denuded human pulmonary arteries; however, DEX had no direct effect on tension in endothelium-intact pulmonary arteries. These findings suggest that the contraction of pulmonary arteries will predominate at plasma concentrations above 10–8 mol/L (approximately 2 ng/ml, which corresponds to the dose for deep sedation) in patients with endothelial dysfunction. We chose serotonin as a contractile stimulus to study the inhibitory effects of DEX on vascular contraction because it has been shown to modulate the functional and remodeling consequences of pulmonary arterial hypertension [27]. Serotonin induced vasoconstriction effectively in our study, which could be attributed to the density of 5-HT receptor subtypes in human pulmonary arteries. Indomethacin had no effect on the inhibitory effect of DEX in the present study, which is opposite to the findings of Wong et al. [6] A similar change in mean pulmonary artery pressure caused by the systemic infusion of DEX at mounting doses in healthy subjects echo the biphasic response as it is shown in the present study.
Most of the adverse events associated with DEX use occur during or briefly after loading of the drug, and blood DEX concentrations may be high enough to cause vasoconstriction immediately after bolus injection [11].
Pekka et al. [28] administered increasing plasma target concentrations of DEX to healthy young volunteers in whom the sympatholytic effects of the drug were attenuated by either general anesthesia or axillary brachial plexus block. The results showed that DEX increased vasoconstriction in anesthetized volunteers, whereas placebo did not. Although the present study was performed in vitro, the peripheral effects of DEX were characterized in isolation from its concurrent sympatholytic effects, which is similar to the changes induced by the systemic infusion of DEX in patients whom the sympatholytic effects of the drug were eliminated as described above.
The present study had one main limitation in that the effects of DEX on pulmonary veins, which contribute significantly to pulmonary vascular resistance, were not measured. Jantschak [29] reported that contractile α2-adrenoceptors play a minor role in porcine pulmonary arteries compared to veins under normal in vitro experimental conditions. Therefore, we suggest that it is premature to conclude that DEX does not affect pulmonary vascular resistance in healthy subjects.
In summary, the present study showed that, in addition to its effect on vasoconstriction, DEX seemed to have a vasodilatory effect through the activation of endothelial NOS in human pulmonary arteries. The relaxing and contracting effects of DEX oppose each other, resulting in little change in tension in quiescent arteries with endothelium. It is plausible that DEX may induce greater pulmonary artery resistance in patients with impaired endothelial function, as in atherosclerosis or diabetes, than in healthy subjects, especially when they are given general anesthesia.
 XML Download
XML Download