

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Alzheimer's disease (AD), a neurodegenerative disease, is one of the most common types of dementia. The mainly pathological features of AD are intracellular neurofibrillary tangles (NFTs) by accumulations of hyperphosphorylated and caspase-truncated tau protein, and extracellular amyloid plaques by aggregations of β-amyloid proteins [1]. The physiological functions of tau are stabilization and assembly of microtubules, regulation of axonal transport and axonal growth [23]. Tau is regulated by posttranslational modifications including phosphorylation. Tau phosphorylation contributes to not only physiological regulation but also tau pathology in tauopathies including AD [4]. Indeed, tau has about 80 residues which are phosphorylation available sites [5], at least 30 residues are phosphorylated by several tau kinase in AD patient's brain [6]. Among tau kinase, glycogen synthase kinase 3β (GSK3β) is known to be a dominant tau kinase which plays an important role in tau pathology [4]. Besides the abnormal and hyper-phosphorylation of tau, caspase cleavage of tau is another factor of tau pathology. The caspase-3 cleaved tau at Asp421 site has been found in NFTs and promotes aggregation of tau [7]. In a previous study, we were confirmed that tau truncated at Asp421 and hyperphosphorylated by GSK3β is more fibrillogenic than wild type tau by sarkosyl fractionation and thioflavin-S staining [8]. These evidences strongly indicate that tau pathologies, hyperphosphorylated and caspase cleaved tau, are involved in progression of AD.
Perturbed calcium homeostasis has been reported to be involved in the progression of AD [910]. Calcium plays important roles in neuron, including synaptic plasticity and apoptosis. Disorders of neuronal calcium signaling have been implicated in the pathogenesis of neurodegenerative disease including AD [11]. Increased intracellular calcium induces the hyperphosphorylation of tau, the accumulation of amyloid-β, and neuronal death. Conversely, Aβ or tau pathology is related with dyshomeostasis of intracellular calcium. Disruption of calcium regulation by ER dysfunction mediates the cellular signaling cascades that are associated with AD [9]. Various stresses, including expression of mutant proteins, accumulation of unfolding and misfolding protein, inflammation, deprivation of glucose, oxygen, or calcium release from of the ER, disrupt ER function and cause so-called ER stress [121314]. Although ER stress initially protects the cell from the toxicity induced by misfolded protein in the ER, it can also cause protein misfolding diseases such as AD [1015]. Several researches reported that the ER stress is activated in the AD brain. In postmortem AD brains, the levels of ER stress markers such as BiP/GRP78 and phosopho-PERK were found to be increased in the cortex and hippocampus. Thapsigargin as an ER-stress inducer stimulated phosphorylation of tau at Thr231, Ser262 and Ser396. Thapsigargin also induced activation of caspase-3 and cleavage of tau [16], suggesting that ER stress may contribute to the tau pathology in AD.
Sorcin, soluble resistance-related calcium-binding protein, is a penta-EF hand calcium binding protein, and highly expressed in the heart, brain, and many cancer cells [17], which regulates intracellular calcium homeostasis by two mechanisms. The major one is the calcium-dependent binding to calcium channels and to other proteins and the other one is calcium binding itself [18].
Although tau pathology, perturbed calcium homeostasis, and ER stress have been suggested to be involved in the progression of AD, the relationship among these factors is not fully elucidated. In the present study, we carefully examined the possible role of pathogenic forms of tau such as GSK3β-induced aberrant phosphorylation and caspase-3 cleavage in the function of calcium binding protein sorcin.
Go to : 
METHODS
Plasmid constructs
Two types of human tau constructs, containing four microtubule binding repeats without exons 2 and 3, were introduced into KpnI and XbaI sites of pcDNA3.1- vector. T4 is full-length and wild type tau. T4C3 is mimicking caspsae-cleaved tau, deleted with last 20 amino acids of c-terminal. GSK3β-S9A is a constitutively active form of GSK3β. T4, T4C3, and GSK-S9A plasmid DNA constructs have been previously described [8]. Full-length human sorcin cDNA was amplified from human liver cDNA library by polymerase chain reaction (PCR) utilizing primers that contained restriction enzyme sites of KpnI and XbaI. The following oligonucleotides were for human sorcin gene amplification; Kpn1-Sorcin (H)-For : 5'-GGGGTACCATG CAGTATGGAGGGGCTCCC-3' and Xba1-Sorcin (H)-Rev : 5'-GCTCTAGATTAAACACTCATGACACATTGAATG-3'. The PCR products were purified using QIAquick PCR Purification Kit by following manufacture's procedure (QIAGEN). Purified PCR products (insert) and pcDNA3.1- (vector) cut off by double digestion with enzymes KpnI and XbaI. After double digestion, insert and vector were electrophoresed on 1.5% (W/V) agarose gel and purified by MEGA-spin™ agarose gel extraction kit (INTRON). The KpnI and XbaI sorcin cDNA fragment was subcloned into pcDNA3.1- vector. All plasmid DNAs were verified by DNA sequencing.
Cell culture
Human embryonic kidney 293 (HEK293) cells and SHSY5Y human nueroblastoma cells were grown in Dulbecco's modified Eagle's medium medium (DMEM) (HyClone, USA) supplemented with 10% fetal bovine serum (HyClone, USA), 1% penicillin/streptomycin (Invitrogen, USA) at 37℃ in humidified incubator with 5% CO2.
Transient Transfections of plasmid DNA and siRNA, and drug treatment
Sorcin siRNA and control siRNA were purchased from Santa Cruz Biotechnology. Plasmid DNA and siRNA were transiently transfected into cells using Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer's protocol. Forty-eight hrs after transfection, the cells were treated 2.5 µM thapsigargin (Sigma) to induce ER stress.
Immunoblotting
Cells were harvested cold PBS, collected by centrifugation, lysed in PRO-PREP protein extraction solution (Intron biotechnology, Korea), and sonicated on ice. Protein concentrations in the supernatants were determined using the bicinchoninic acid assay (Sigma). Equal amounts of proteins were diluted with 2X protein loading buffer (0.25 M Tris-HCl, pH 6.8, 5 mM EDTA, 5 mM EGTA, 25 mM dithiothreitol, 2% SDS, and 10% glycerol with bromophenol blue as the tracking dye), incubated in a boiling water bath for 5 min, separated on 8~15% SDS-polyacrylamide gels or gradient gels, transferred to PVDF membrane (GE Healthcare), and blots were blocked in 5% nonfat dry milk in TBST (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.05% Tween 20) for 30 min at room temperature, and probed with indicated antibodies in same buffer overnight at 4℃. The membranes were then washed with TBST and incubated with HRP-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch) or HRP-conjugated goat anti-mouse IgG (Jackson ImmunoResearch) for 2 hr at room temperature. The membranes were rinsed with TBST. The blots were visualized with enhanced chemiluminescence (GE Healthcare) and exposed on film (Kodak). Gel images were scanned and quantified using Image Quant software (Molecular Dynamics, Sunnyvale, California, USA). For fair comparison, β-actin was used as a loading control. The following antibodies were used in the present study; anti-5A6 for total tau (phosphorylation-independent) (Biomax); anti-BiP antibody (Abcam) for ER stress maker; anti-cleaved PARP and anti-cleaved caspase-12 antibodies (Cell Signaling) to detect apoptosis; anti-sorcin (Santa Cruz); anti-β-actin antibody (Sigma).
Immunoprecipitation
Agarose beads, protein A Sepharose® and Protein G Sepharose ®, were purchased from Sigma. Cell lysates containing 200 µg of protein were precleared for 2 hr at 4℃ with agarose beads on gentle rotation. Precleared samples were immunoprecipitated overnight at 4℃ with the tau polyclonal antibody (Dako) or sorcin monoclonal antibody (Santa cruz). Agarose beads were added and the incubation continued for 2 hr at 4℃. After centrifuge at 14,000×g for 1 min at 4℃, the supernatant was removed and the precipitated agarose beads were collected and washed three times with PRO-PREP protein extraction solution, and 40 µl of 2X reducing stop buffer was added to each sample and the samples were placed in a boiling water bath for 15 min before SDS-PAGE and immunoblotting.
Immunocytochemistry
HEK 293 cells were seeded on coverslips in 6-well plates. After 24 hr of seeding, the cells were transiently transfected T4 or T4C3. Forty-eight hrs later, cells were fixed with 3% PFA (paraformaldehyde) for 15 min at RT, and washed by PBS three times. For permeabilization, the cells were incubated with 0.2% Triton X-100 in PBS for 2 min, and washed. To reduce background, cells were blocked with 4% BSA (bovine serum albumin) in PBS for 30 min. Afterwards, the cells were incubated with goat polyclonal sorcin antibody (Santa Cruz) for overnight at 4℃ and rabbit polyclonal tau antibody (Dako) for 2 hr at RT. Localizations were visualized using FITC-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch), and Alexa Fluor 568-conjuated donkey anti-goat IgG (Invitrogen) secondary antibody. For staining nucleus, the cell were incubated with 5 µg/ml Hoechst 33258 (Sigma) for 1 hr at RT, rinsed by PBS three times, and mounted. The images were taken by using a confocal laser microscope (Olympus; FV10-ASW, Fluoview).
Measurement of intracellular Ca2+ levels
HEK 293 cells were seeded on round coverslips in 6-well plates. After 24 hr of seeding, the cells were transiently transfected tau constructs with / without sorcin or sorcin siRNA. Forty-eight hr later of transfection, the cells were incubated with 2 µM Fluo-4AM (Invitrogen) for 30 min at 37℃ in humidified incubator. Each coverslip containing stained cells were scanned by confocal laser microscope with a 488-nm excitation argon laser and a 510-nm long pass emission filter. The Flou-4 AM fluorescence intensities were taken using Fluoview FW1000 software (Olympus) by randomly selected 30 cells from three independent experiments.
Statistical analysis
Data were expressed as mean±SD, obtained from at least three independent experiments, and analyzed using SPSS 12.0 statistical software (SPSS Inc., USA). The one-way analysis of variance (ANOVA) procedure followed by Tukey's post hoc tests was used to analyze the differences among groups. Values of p<0.05 were considered statistically significant.
Go to :
RESULTS
Tau interacts with sorcin and expression of caspase-3-cleaved tau potentiates tau-sorcin interaction
In the previously study, we have demonstrated that caspase-3-cleaved tau interacts with sorcin using yeast two hybrid system [19]. To confirm the interaction of tau with sorcin in cells, we performed co-immunoprecipitation assay using HEK293 cells and SH-SY5Y cells after transient transfection of constructs. Cells were transfected with T4, T4C3, or vector. After 48 hr of transfection, cells were harvested. Cell lysates were immunoprecipitated with tau or sorcin antibody, and detected with respective antibodies in Western blotting. Empty vector of pcDNA3.1- was used for transfection control and agarose beads for immunoprecipitation. The result of immunoprecipitation assay showed that tau was directly interacted with sorcin in both HEK293 and SH-S5Y cells (Fig. 1A and B). Especially, T4C3 showed significantly increased interaction of tau and sorcin compared to T4 (Fig. 1A and B). Previously, it has been reported that sorcin localizes in ER-derived vesicles along the microtubules [20], suggesting a possibility of interaction with tau. Immunocytochemistry was carried out to examine the intracellular localization of tau and sorcin. Both tau and sorcin were diffuse in cytoplasm. Especially, sorcin appeared to be highly localized in peri-nuclear areas. Sorcin co-localized with both T4 and T4C3 tau. However, the occurrence of colocalization was obviously more observed in T4C3 compared to T4 (Fig. 1C), which is consistent with immunoprecipitation data. T4C3 cells appeared abnormal in morphology compared to T4 cells. It has been reported that tau is a substrate of caspase-3 and that caspase cleavage of tau is an early event of NFT formation, which subsequently accompanies morphologic changes including shrinkage [21].
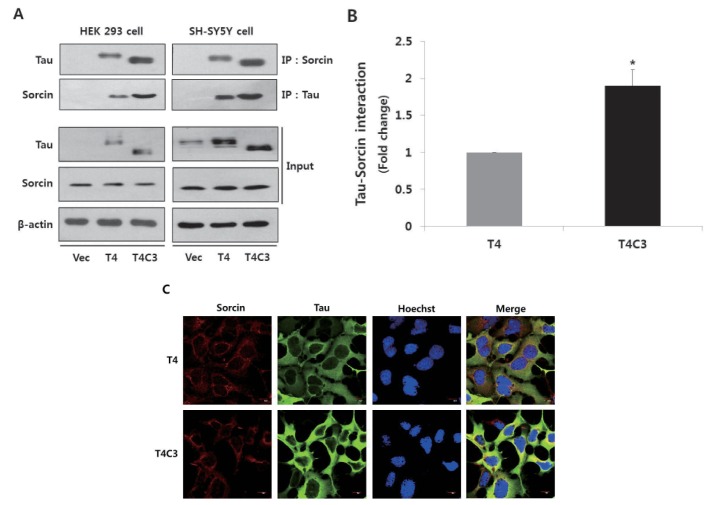 | Fig. 1Tau interacts with sorcin.Representative images (A) and quantitative analysis (B) of immunoprecipitation of tau and sorcin. Intracellular localization of tau and sorcin (C). HEK 293 and SH-SY5Y cells were transfected with T4, T4C3, or vector. After 48 hr of transfection, cells were harvested. Cell lysates were immunoprecipitated with tau or sorcin antibody, and detected with Western blotting with respective antibodies. Vector was used for transfection control and agarose beads control in immunoprecipitation data to conform binding of agarose beads to tau or sorcin. Data were obtained from independent experiments (n=3) and expressed as mean±SD. *p<0.05 indicates significant differences between the indicated groups. To examine the intracellular localization of tau and sorcin, T4 or T4C3 transfected HEK 293 cells were stained with sorcin (red) or tau (green) antibody. Nuclei were visualized by Hoechst staining (blue). Scale bar 10 µm. Vec stands for vector (pcDNA3.1-) and IP for immunoprecipitation.
|
Phosphorylation of tau by GSK3β and ER stress by thapsigargin potentiate interaction of tau with sorcin
Hyperphosphorylation of tau by GSK3β, truncation of tau by caspase-3, and activation of ER stress are involved in the pathogenesis of tauopathies. To investigate the effect of these factors on tau-sorcin interaction, cells were co-transfected each tau constructs with GSK3β or vectors. After 48 hrs of transfection, cells were treated 2.5 µM thapsigargin for 24 hr to induce ER stress. Thapsigargin, an inhibitor of sarcoplasmic-endoplasmic reticulum Ca2+ ATPase, is well characterized as an inducer of ER stress and excessive treatments of thapsigargin lead to cell death [22]. As shown in Fig. 1, T4C3 cells exhibited significantly increased tau-sorcin interaction compared to T4 cells. In T4 cells, tau-sorcin interaction was potentiated with the treatment of thapsigargin (T4-Vec-Tg) and co-expression of GSK3β (T4-GSK3β-Cont) compared to T4-Vec-Cont, respectively. Also, expression of GSK3β with treatment of thapsigargin (T4-GSK3β-Tg) in T4 cells remarkably increased tau-sorcin interaction (Fig. 2A and B). In T4C3 cells, no significant difference was observed with thapsigargin treatment, although a similar pattern with T4 cells was observed (Fig. 2A and B). The expression of BiP, an ER chaperon protein and ER stress maker, was examined in the present study. BiP was significantly increased with thapsigargin treatment in both T4 and T4C3 cells (Fig. 2A and C). However, significant potentiation of BiP expression was observed in T4C3 cells with the treatment of thapsigargin compared to T4 cells (Fig. 2A and C). These data strongly suggest that sorcin preferentially associates with fibrillogenic tau species such as GSK3β-induced hyperphosphorylated and caspase-3-cleaved tau.
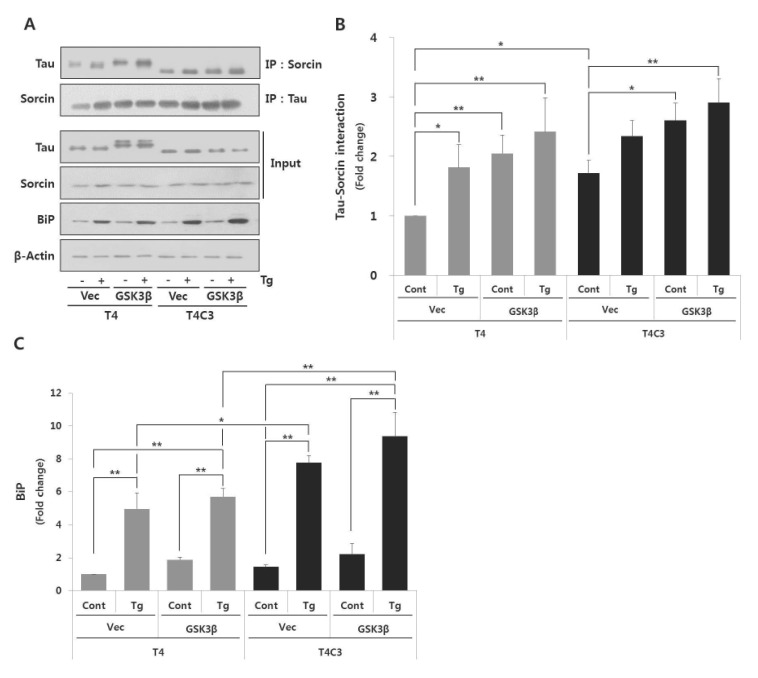 | Fig. 2Fibrillogenic tau species preferentially associates with sorcin and increases ER stress induced by thapsigartin compared to wild type tau.Immunoprecipitation of tau and sorcin (A). Quantitative analyses of Tau-sorcin interaction (B) and bip expression (C). HEK 293 cells were co-transfected T4 or T4C3 with GSK3β-S9A or vector, respectively. After 48 hr of transfection, cells were treated 2.5 µM thapsigartin for 24 hr to induce ER stress. Cell lysates were immunoprecipitated with tau or sorcin antibody, and performed Western blotting. BiP was used as an ER stress maker. Data were obtained from independent experiments (n=3) and expressed as mean±SD. *p<0.05 and **p<0.01 indicate significant differences between the indicated groups. Vec stands for vector (pcDNA3.1-), cont for control, Tg for thapsigargin, and IP for immunoprecipitation.
|
Expression of caspase-3-cleaved tau increases thapsigargin-induced cytotoxicity
To examine the effect of interactions on thapsigargin-induced cytotoxicity, cell viability was measured during 48 hr after treatment of thapsigargin by MTT assay. No significant difference was observed in the absence of thapsigargin in both T4 and T4C3 cells. However, T4C3 cells showed significantly decreased cell viability with the treatment of thapsigargin compared to T4 cells (Fig. 3A and C). Thapsigargin-induced cytotoxicity was significantly potentiated in groups showing increased tau-sorin interactions (Fig. 3), suggesting that thapsigargin-induced cytotoxicity was dependent on tau-sorcin interaction. To confirm the role of sorcin in ER stress-induced cytotoxicity, sorcin expression levels were modulated using the transient transfection of sorcin overexpression plasmid or sorcin siRNA. Overexpression of sorcin significantly attenuated thapsigargin-induced cytotoxicity in T4C3 cells, whereas silencing of sorcin by sorcin siRNA potentiated thapsigargin-induced cytotoxicity in T4 cells (Fig. 3).
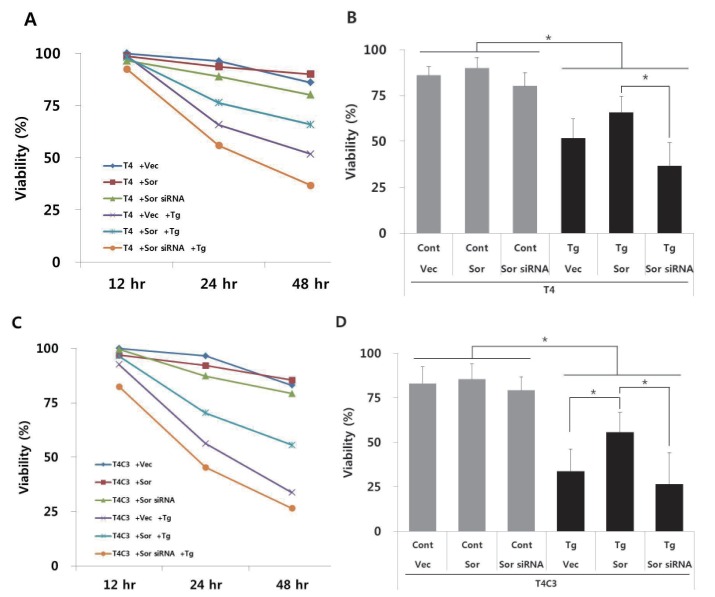 | Fig. 3Expression of caspase-3 cleaved tau and knock-down of sorcin increase thapsigartin-induced cytotoxicity but over-expression of sorcin decreases thapsigartin-induced cytotoxicity.HEK293 cells were transiently transfected with T4 (A, B) or T4C3 (C, D) constructs in the absence or presence of sorcin overexpression or sorcin siRNA. After 48 hrs of transfection, cells were treated 2.5 µM thapsigartin. Cell viability was measured with MTT assay (A, C) and statistical analyses of cell viability were determined at 48 hr after thapsigartin treatment (B, D). Data were obtained from independent experiments (n=3) and expressed as mean±SD. *p<0.05 and **p<0.01 indicate significant differences between the indicated groups. Vec stands for vector (pcDNA3.1-), Sor for sorcin, Cont for control, and Tg for thapsigargin.
|
Overexpression of sorcin attenuates thapsigargin-induced apoptosis whereas knock-down of sorcin accelerates thapsigargin-induced apoptosis
To further confirm the role of sorcin in ER stress-induced apoptosis, the levels of apoptotic markers such as PARP and caspase-12 were examined. Both genes are well characterized apoptosis-involved proteins; PARP, as a marker of apoptosis [23] and caspase-12, as ER-stress mediated apoptosis caspase [24]. As shown in Fig. 2B and 3, thapsigargin treatment exhibited significantly increased cytotoxicity and BiP expression in both T4 and T4C3 cells. Also, increased BiP expression and PARP and caspase-12 cleavages by thapsigargin were observed in T4C3 cells compared to T4 cells (Fig. 4 and 5). Above all, overexpression of sorcin significantly attenuated thapsigargin-induced apoptosis in T4C3 cells (Fig. 3 and 4), whereas silencing of sorcin potentiated thapsigargin-induced apoptosis in T4 cells (Fig. 3 and 5). These results strongly suggest that unbound free sorcin might play an important role to resist the thapsigargin-induced ER stress. Overexpression of sorcin significantly attenuated thapsigargin-induced apoptosis in T4C3 cells (Fig. 4). In T4C3 cells, considerable amount of sorcin was sequestered to T4C3. Therefore, unbound amount of sorcin might not be sufficient to defend the cells against thapsigargin-induced ER stress. However, in T4 cells, there are abundant free sorcin due to the fact that T4 does not sequester sorcin. Therefore, there is no significant difference by overexpression of sorcin in thapsigargin-induced apoptosis in T4 cells (Fig. 4). Reversely, knock-down of sorcin significantly increased thapsigargin-induced apoptosis in T4 cells, whereas no noticeable potentiation was observed in T4C (Fig. 5). These data strongly indicate that availability of unbound free form of sorcin might be important to defend against the thapsigargin-induced ER stress.
 | Fig. 4Overexpression of sorcin significantly attenuates thapsigartin-induced apoptosis in T4C3 cells.Representative immunoblots (A) of examined proteins and quantitative analyses of cleaved PARP (B) and cleaved caspase-12 (C). HEK293 cells were transiently transfected T4 or T4C3 constructs and then co-transfected with sorcin or vector, respectively. After 48 hrs of transfection, cells were treated 2.5 µM thapsigartin for 24 hr. Western blotting was carried out with cleaved PARP and cleaved caspase-12 antibodies to determine role of sorcin on thapsigartin-induced apoptosis. Data were obtained from independent experiments (n=3) and expressed as mean±SD. *p<0.05 and **p<0.01 indicate significant differences between the indicated groups. Vec stands for vector (pcDNA3.1-), Sor for sorcin, Cont for control, and Tg for thapsigargin.
|
 | Fig. 5Knock-down of sorcin significantly accelerates thapsigartin-induced apoptosis in T4 cells.Representative immunoblots (A) and quantitative analyses of cleaved PARP (B) and cleaved caspase-12 (C). HEK293 cells were transiently transfected T4 or T4C3 constructs and then co-transfected with sorcin siRNA or control siRNA, respectively. After 48 hr of transfection, cells were treated with 2.5 µM thapsigartin for 24 hr. Western blotting was carried out with cleaved PARP and cleaved caspase-12 antibodies to determine role of sorcin on thapsigartin-induced apoptosis. Data were obtained from independent experiments (n=3) and expressed as mean±SD. **p<0.01 indicates significant differences between the indicated groups. Vec stands for vector Sor for sorcin, Cont for control, and Tg for thapsigargin.
|
Expression of caspse-3-cleaved tau and silencing of sorcin potentiate the thapsigargin-induced perturbation of intracellular calcium homeostasis
To examine the calcium buffering ability of sorcin and the effect of tau-sorcin interaction on calcium buffering capacity, intracellular calcium concentration was examined using Flou-4AM staining with confocal microscope at 48 hr after transient transfection of respective constructs. Dysregulation of calcium signaling has been implicated in the pathogenesis AD [11], and treatment of thapsigargin has been reported to result in a global and transient increase in cytosolic calcium levels [2526]. Intracellular calcium concentration was rapidly increased by thapsigargin both T4 and T4C3 cells (Fig. 6). Peak in intracellular calcium concentrations was reached in 400 sec in almost groups. T4C3 cells exhibited higher thapsigargin-induced intracellular calcium concentration compared to T4 cells. Knock-down of sorcin showed significantly increased thapsigargin-induced perturbation of intracellular calcium homeostasis in both cells. This pattern is maintained until the end of the examination (Fig. 6). The data is in accordance with the apoptosis results. These data strongly suggest that T4C3, caspase-cleaved tau, results in the significantly compromised function of sorcin such as intracellular calcium homeostasis through the sequestration of sorcin into fibrillogenic tau.
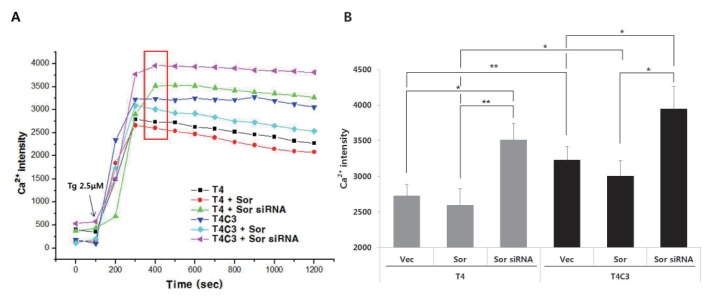 | Fig. 6Expression of caspse-3-cleaved tau and silencing of sorcin potentiate the thapsigargin-induced perturbation of intracellular calcium homeostasis.Representative image (A) and quantitative analysis (B) of intracellular calcium levels. Cultured cells on coverslips were co-transfected with indicated constructs. After 48 hr transfection, cells were incubated with 2 µM Flou-4AM for 30 min to stain intracellular calcium. Data were obtained from randomly selected 30 cells from three independent experiments (n=3) and expressed as mean±SD. *p<0.05 and **p<0.01 indicate significant differences between the indicated groups. Vec stands for vector (pcDNA3.1-) and Sor for sorcin.
|
Go to :
DISCUSSION
In a previous study, we identified that sorcin interacts with caspase-3 cleaved tau in yeast two hybrid analysis using caspase-3-cleaved tau [19]. However, the consequence of tau-sorcin interaction still remained unexplained. Base on yeast two hybrid screening data, we first examined whether tau directly interacts with sorcin with full length tau (T4) and caspase-3-cleaved tau (T4C3) using co-immunoprecipitation analysis. Tau was directly interacted with sorcin, and T4C3 cells were showed increased interaction of tau with sorcin compared to T4 cells (Fig. 1). T4C3, caspase-3-cleaved tau, has been reported to be highly fibrillogenic with a potential to progress to the NFT [2526]. NFTs are one of the primary pathological hallmarks of AD and have been suggested to play a major role in facilitating neuronal degeneration. NFTs are mainly composed of paired helical filaments (PHFs), which are formed from abnormally hyperphosphorylated and cleaved tau accumulation [27]. Aberrant phosphorylation and caspase cleavage have been reported to play a role in the aggregation of tau [72829]. Tau in NFTs has been shown to be truncated at Asp421 [1730]. Truncated tau, albeit not a large amount, may play a significant role in the neuronal cell death and PHF formation, given that truncated tau has been shown to be associated with apoptosis in cultured cells [293132], and has been demonstrated to play a significant role in the nucleation-dependent filament formation of tau [20]. Therefore, it is important to investigate how GSK3b-mediated phosphorylation and caspase cleavage of tau contribute to the initiation of the tau aggregation process. In a previous study, it was demonstrated that tau truncated at Asp421 (T4C3), but not full-length tau (T4), partitioned into a sarkosyl-insoluble fraction and formed thioflavin S-positive aggregates when coexpressed with GSK3β [33]. This indicates that a combination of phosphorylation and cleavage of tau may coordinate in regulating tau aggregation. These evidences suggest that hyperphosphorylation and truncation of tau are important factors of taupathology. Along with tau pathology, ER stress has been reported to be related in AD [1015]. Thapsigargin, as an ER-stress inducer, induces phosphorylation of tau, activation of caspase, and cleavage of tau [16]. So, we examined the possible effect ER stress by thapsigargin on tau-sorcin interaction. As shown in Fig. 2, co-transfection GSK3β with tau constructs and treatment of thapsigargin increased the interaction of tau with sorcin, indicating that fibrillogenic tau species preferentially associates with sorcin.
Disorders of neuronal calcium signaling have been implicated in neurodegenerative disease including AD [11]. Calcium binding protein sorcin is highly expressed in brain, about 5~10 times higher than in heart. Sorcin participates in the regulation of intracellular calcium homeostasis through two different mechanisms. The first one is calcium binding itself; sorcin binds calcium up to micromolar levels [3435]. The second one, the major mechanism, is the calcium-dependent binding to calcium channels and to other proteins. Sorcin is able to regulate L-type calcium channel and Na+–Ca2+ exchangers (NCX) in plasma membrane, ryanodine receptor (RyR) and sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) in ER by interaction with them [18363738]. Also, sorcin is over-expressed in many human tumors, which is involved in the resistance to chemotherapeutic agents in cancer cells. Sorcin expression increases calcium concentration in ER, can prevent ER stress and the unfolded protein response, and increases the resistance from apoptosis [3940]. Conversely, sorcin silencing activates apoptotic proteases such as caspase-3, caspase-12 and ER chaperones such as GRP78/BiP [39]. Consequently, sorcin silencing results in significant defects in mitosis and cytokinesis, blocks cell cycle progression in mitosis, increases the number of rounded polynucleated cells, and induces apoptosis and cell death [21]. These findings suggest that the magnitude of tau-sorcin interaction could affect functions of sorcin such as apoptosis and intracellular calcium regulation. To test this hypothesis, we investigated thapsigargin-induced cytotoxicity and calcium buffering ability. Interaction-dependently, T4C3 cells showed potentiated effect with thapsigargin than T4 cells in cytotoxicity (Fig. 3) and in intracellular calcium dysfunction (Fig. 6). These data strongly suggest that increased tau-sorcin interaction significantly compromises functions of sorcin. To identify the role of sorcin in ER stress-induced apoptosis, we modulated sorcin expression levels with transient transfections of sorcin construct or sorcin siRNA. Interestingly, overexpression of n b sorcin significantly attenuated thapsigargin-induced apoptosis in T4C3 cells, whereas silencing of sorcin accelerated thapsigargin-induced apoptosis in T4 cells (Fig. 4 and 5). There are some evidences that sorin is related with neurodegenerative disease. Sorcin is directly interacts with with presenilin 2 (PS2) and α-synuclein by calcium dependent manner [4142]. PS2 is involved in pathogenesis of AD, which regulates amyloid precursor protein (APP) processing as a gamma-secretase. Also, mutations in presenilin 1 (PS1) and PS2 are associated with early-onset familial Alzheimer's disease (FAD) [43]. Aggregation of α-synuclein is well known as feature of Parkinson's diseases. α-Synuclein pathology is also found in both familial and sporadic case with AD [44]. Moreover, α-synuclein is a interacting protein with tau [45]. These evidences strongly support that tau interacts with sorcin, which may result in the loss of sorcin function such as homeostasis of intracellular calcium and resistance to ER stress.
In the present study, we demonstrated that interaction of tau with sorcin significantly increased with thapsigargin-induced ER stress and fibrillogenic tau species such as GSK3β-induced hyperphosphorylated and caspase-cleaved and that sequestration of sorcin by aberrant tau species resulted in the impairment of sorcin functions such as calcium homeostasis and ER stress resistance. These data strongly suggest that loss of sorcin functions by increased tau-sorcin interactions might play important roles in initiation and/or progression of tau pathology in AD.
Go to :
 XML Download
XML Download