

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pain is associated with a wide range of injury and disease. Worldwide, more than 1.5 billion people suffer from acute or chronic pain, as reported by Global Industry Analysts. Global Burden of Disease (GBD) study shows that burdens associated with chronic pain are increasing in many types of diseases and injuries [1]. In addition, pain causes depression and vice versa. There is a close relationship between pain and depression. Antidepressant medications help relieve pain and depression [2]. Numerous approaches for the treatment of chronic pain have been investigated, but the management of chronic pain with the available pharmacologic therapies is often unsatisfactory. New therapeutic approaches for the prevention and treatment of chronic pain are needed.
A large number of studies have shown that ion channels are closely connected with chronic pain [345]. TWIK-related K+ channel-2 (TREK-2) and TWIK-related spinal cord K+ (TRESK) channel are members of two-pore domain K+ (K2P) channel. These channels are the main contributors to the background current in dorsal root ganglion (DRG) neurons [67]. TREK-2 is involved in mechanical and osmotic pain and in cold allodynia [8]. In addition, TREK-2 expressed in IB4-binding C-fiber nociceptors limits spontaneous pain [9]. TRESK channels activated by the inflammatory mediator (lysophosphatidic acid) reduce nociceptive signaling in DRG neurons [10]. TRESK is associated with migraine [3711]. Familial migraine with aura is associated with a dominant-negative mutation in human TRESK channels. A dominant-negative mutation in TRESK induces hyperexcitability when expressed in TG neurons [11]. TRESK knockdown mice show increased pain and sensitivity in response to painful stimuli. Thus, a prominent physiological role of TREK-2 and TRESK has been attributed to pain sensation [12]. Therefore, an up-regulation of TREK-2 and TRESK channel activities may be a useful strategy in the development of new therapies for the treatment of many types of pain. Activators of the TREK and TRESK channel are expected to emerge as a novel class of analgesic agents [13].
Non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen and nabumetone, are widely used for pain relief. In addition to classical analgesics, antidepressants are useful therapeutic strategies for pain [131415]. Amitriptyline, bupropion, and fluoxetine produced antiallodynic effects in chronic constriction injured mice [16]. Of antidepressants, tricyclic antidepressants (TCAs) are the most established drugs in the treatment of chronic pain [1718], but selective serotonin reuptake inhibitors (SSRIs) or selective norepinephrine reuptake inhibitors (SNRIs) are also effective against pain [1819].
A better understanding of the relationships between these drugs and the changes they induce in TREK-2 and TRESK channels should help to identify more effective treatments for pain. This study was performed to identify the effect of analgesics that are currently used in clinics on TREK-2 and TRESK channels.
Go to : 
METHODS
Chemicals
All of the chemicals used in this study were purchased from Sigma Chemical Company (St. Louis, MO, USA) unless otherwise specified. Stock solutions of escitalopram (20 mM), fluoxetine (100 mM), and ibuprofen (100 mM) were prepared in dimethyl sulfoxide (DMSO) and then diluted in experimental solution to a working concentration. Acetaminophen (500 mM) was dissolved in ethanol. Amitriptyline (100 mM), bupropion (100 mM), citalopram (10 mM), and nabumetone (100 mM) were dissolved in distilled water. When DMSO or ethanol was used as a solvent, a solution containing an equivalent concentration was used as a control.
Transfection
HEK293 cells were seeded at a density of 2×105 cells per 35 mm dish 24 h prior to transfection in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS. HEK293 cells were co-transfected with DNA fragments encoding mouse TRESK (NM_207261) or rat TREK-2 (NM_023096) and green fluorescent protein (GFP) in pcDNA3.1 using LipofectAMINE2000 and OPTI-MEM I Reduced Serum Medium (Life technologies, Grand Island, NY, USA). Green fluorescence from cells expressing GFP was detected with the aid of a Nikon microscope equipped with a mercury lamp light source. Cells were used 2~3 days after transfection.
Electrophysiological studies
Electrophysiological recording was performed using a patch clamp amplifier (Axopatch 200, Axon Instruments, Union City, CA, USA). Pipette tip resistances were 4~6 MΩ. Wholecell current was recorded in response to a voltage ramp (–120 to +60 mV; 865 ms duration) from a holding potential of –80 mV in physiological solution containing 5 mM KCl. Currents were filtered at 2 kHz, and the currents measured at +60 mV were obtained and analyzed. Bath solution contained (mM): 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 5 glucose and 10 HEPES (pH 7.4), and pipette solutions contained (mM): 150 KCl, 1 MgCl2, 5 EGTA and 10 HEPES (pH 7.3). The pH was adjusted to desired values with HCl or NaOH. All experiments were performed at ~25℃.
Statistical analysis
Differences among groups were analyzed using one-way ANOVA test (SPSS18 software, SPSS, Chicago, IL, USA). Data are represented as mean±SD. A p<0.05 was considered as the criterion for significance.
Go to :
RESULTS
Comparison of effect of analgesics on TREK-2 and TRESK overexpressed in HEK-293 cells
Application of the NSAIDs (ibuprofen and nabumetone) and acetaminophen to TREK-2 and TRESK channels that were overexpressed in HEK-293 cells produced different effect in TREK-2 and TRESK currents. As shown in Fig. 1, acetaminophen (100 µM), ibuprofen (100 µM), and nabumetone (100 µM) inhibited TRESK by 27±7%, 21±9%, and 32±11%, respectively, but they did not affect TREK-2. The IC50 for inhibition of TRESK by acetaminophen, ibuprofen, and nabumetone was 220±26 µM, 889±10 µM, and 557±35 µM.
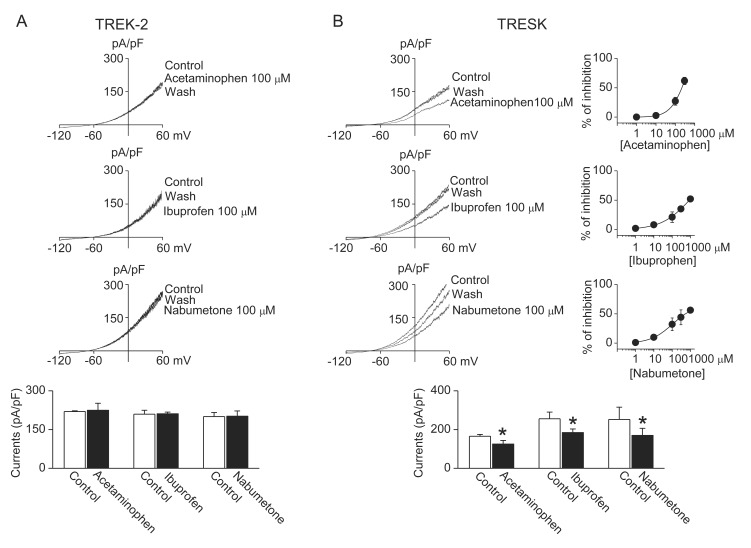 | Fig. 1Comparison of effect of analgesics on TREK-2 and TRESK.Wholecell currents were recorded in 5 mM KCl, and the current levels at +60 mV were determined and analyzed. (A) No effect of acetaminophen, ibuprofen, and nabumetone on TREK-2 currents. (B) Inhibitory effect of acetaminophen, ibuprofen, and nabumetone on TRESK currents. Dose-response curves of analgesics are shown on the right of representative current traces. The bar graphs show the effect of analgesics on TREK-2 and TRESK currents. Data represent the mean±SD of five repeated experiments. *p<0.05 compared to the corresponding control.
|
Antidepressants used in this study, except bupropion, inhibited both TREK-2 and TRESK currents. Amitriptyline (a TCA, 30 µM) inhibited TREK-2 and TRESK currents by 71±10% and 54±16%, respectively. The inhibitory effects of amitriptyline were higher on TREK-2 current than on TRESK current. The IC50 for inhibition of TREK-2 and TRESK by amitriptyline was 16±4 µM and 26±5 µM, respectively (Fig. 2A). Bupropion, a specific dopamine reuptake inhibitor, had no effect on TREK-2, but inhibited TRESK currents by 30±17%. The IC50 for inhibition of TRESK by bupropion was 160±17 µM (Fig. 2B). Bar graphs show the effect of amitriptyline and bupropion on TREK-2 and TRESK. Also, SSRIs (fluoxetine, citalopram, and escitalopram) inhibited both TREK-2 and TRESK currents. Citalopram, escitalopram, and fluoxetine inhibited TREK-2 currents by 49±10%, 55±12%, and 71±8% at the concentration of 100 µM (Fig. 3A). Citalopram, escitalopram, and fluoxetine also inhibited TRESK currents by 30±6%, 72±7%, and 92±10%, respectively (Fig. 3B). The IC50 for inhibition of TREK-2 by citalopram, escitalopram, and fluoxetine was 110±9 µM, 102±9 µM, and 31±7 µM, respectively. The IC50 for inhibition of TRESK by citalopram, escitalopram, and fluoxetine was 168±11 µM, 49±7 µM, and 17±5 µM, respectively. The inhibitory effect of escitalopram and fluoxetine was higher on TRESK current than on TREK-2 current. The inhibitory effect of citalopram on TREK-2 was similar to that on TRESK. The currents recorded from GFP-transfected cells were ~10 pA/pF. All drugs had no effect on the currents (data not shown).
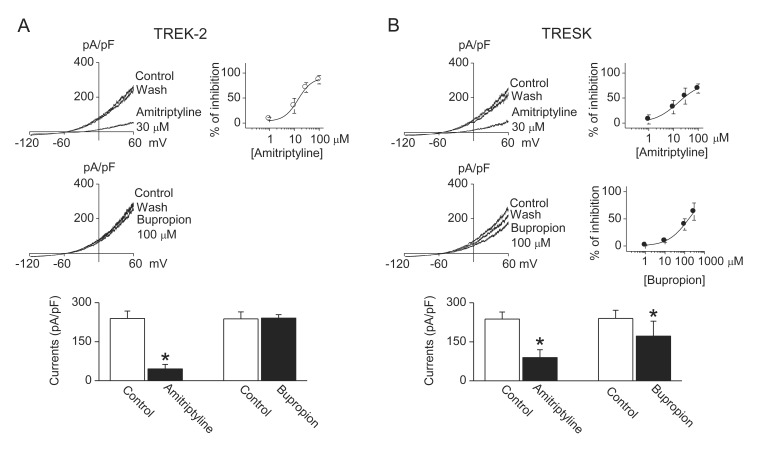 | Fig. 2Inhibition of TREK-2 and TRESK currents by amitriptyline.(A and B) Effect of amitriptyline and bupropion on TREK-2 and TRESK currents. Doseresponse curves of amitriptyline and bupropion are shown on the right of representative current traces. The bar graphs show the effect of amitriptyline and bupropion on TREK-2 and TRESK channels. Data represent the mean±SD of five repeated experiments. *p<0.05 compared to the corresponding control.
|
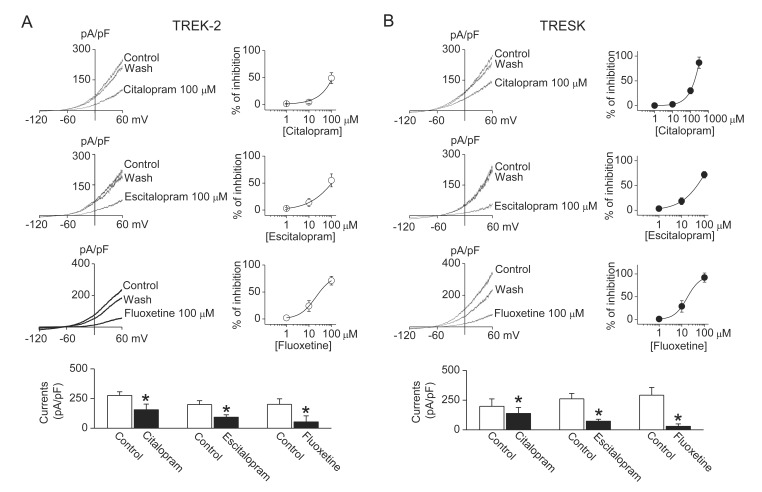 | Fig. 3Inhibition of TREK-2 and TRESK currents by SSRIs antidepressants.(A and B) Inhibitory effect of citalopram, escitalopram, and fluoxetine on TREK- 2 and TRESK currents. Dose-response curves of SSRI antidepressants are shown on the right of representative current traces. The bar graphs show the effects of analgesics on TREK-2 and TRESK. Data represent the mean±SD of five repeated experiments. *p<0.05 compared to the corresponding control.
|
Go to :
DISCUSSION
This study reports the effects of therapeutic compounds for pain on TREK-2 and TRESK currents. We hypothesized that analgesics commonly used in clinics may activate TREK-2 and TRESK, because a loss-of function in TREK-2 and TRESK induces depolarization and results in pain [78910]. However, the most of analgesics tested in this study inhibited TREK-2 and TRESK currents. These compounds included amitriptyline, citalopram, escitalopram, and fluoxetine. Acetaminophen, bupropion, ibuprofen, and nabumetone showed different effect on TREK-2 and TRESK; they inhibited TRESK currents, but had no effect on TREK-2.
The concentrations of compounds used in this study were lower than adult dose, as reported by U.S. Food and Drug Administration (FDA), but slightly higher than their blood concentrations. However, in the case of acetaminophen (15 µg/mL), ibuprofen (20 µg/mL), and nabumeton (22.8 µg/mL), the concentrations were similar to their blood concentrations ranging 5 to 20 µg/mL [20], 8.4 µg/mL when receiving 400 mg/day ibuprofen [21], and 12~16 mg after administration of 2000 mg/day nabumetone (U.S. FDA, NDA 19-538/S-023), respectively. The dose of ibuprofen can be increased to 1200 mg/day. The blood concentration of nabumetone is calculated with its principal active metabolite 6-methoxy-2-naphthylacetic acid (6MNA) detected in the blood after administration of nabumetone. The blood concentrations of amitriptyline, bupropion, citalopram, escitalopram, and fluoxetine are 0.34 µg/ mL, 818±500 ng/mL, 49 ng/mL, 11.6~46.4 ng/mL, and 97±51 ng/mL after oral administration of 10 mg/day [22], 252 mg/ day [23], 20 mg/day citalopram [24], 5~40 mg/day [25], and 20 mg/day [26], respectively. Their blood concentrations were lower than concentrations used in this study (amitriptyline (8.4 µg/mL), bupropion (23.9 µg/mL), citalopram (32.4 µg/mL), escitalopram (41.4 µg/mL), and fluoxetine (30.9 µg/mL)). The low blood concentration of antidepressants will not be likely to inhibit TRESK and TREK-2 currents in vivo. However, the dose can be increased depending on symptom improvement. In addition, the concentrations of drugs are different among organs (e.g. fluoxetine concentration is different between blood and brain; the concentration is higher in brain than in blood [27]). Acetaminophen, ibuprofen, and nabumetone have a possibility that they affect TRESK in in vivo.
Acetaminophen and NSAIDs could modulate K+ channels. NSAIDs activate the Kv7 channel and large-conductance Ca2+-activated K+ channel in vascular smooth muscle cells [2829]. In contrast, NSAIDs inhibit the surface expression of Kv1.4 and Kv1.6 and depolarize membrane potentials in intestinal epithelial cells [30]. Acetaminophen protects estramustineinduced cytotoxicity on cultured fibroblast by blocking inhibition of cellular K+ channel ion transport [31]. These drugs affect K+ channels expressed in different type of cells differently. Acetaminophen, ibuprofen, and nabumetone can be used to distinguish TREK-2 and TRESK expressed in sensory neurons. Further study will be needed to identify the mechanism by which acetaminophen, ibuprofen, and nabumetone modulate TREK-2 and TRESK current differently.
Antidepressants, which have analgesic effects, modulate many types of K+ channels. Within K2P channel family, TREK- 1 and TASK-3 have been suggested as potential targets for antidepressants [323334]. These channels are inhibited by fluoxetine. Our previous study reported that TREK-2 and TRESK currents were inhibited by fluoxetine [35,36]. The present study demonstrates that TREK-2 and TRESK are also inhibited by other antidepressants, such as amitriptyline, citalopram, and escitalopram. Bupropion showed different effect on TREK- 2 and TRESK. Bupropion, which inhibits the reuptake of norepinephrine and dopamine, improves pain relief [37]. In addition, bupropion interacts with nicotinic receptor [38]. Bupropion blocks KATP channel as well as TRESK [39]. TREK- 2 and TRESK show different effect in response to some signals. Acetylcholine (carbachol) inhibits TREK-2 currents, but activates TRESK current [354041]. Aristolochic acid, a traditional medicine used in the treatment of pain, activates TREK-2, but inhibits TRESK activity [42]. TREK-2 channels are activated by low intracellular pH (pHi) and arachidonic acid, and inhibited by PKC activation [4043]. In contrast, TRESK channels are inhibited by low pHi and arachidonic acid [44], and human (not murin) TRESK is activated by PKC [45]. In this study, we tested effects of analgesics and antidepressants on rat TREK-2 and mouse TRESK. Species difference could explain the different effects of the drugs on TREK-2 and TRESK. In addition, the state of phosphorylation is different between TREK-2 and TRESK. TRESK is constitutively phosphorylated under resting conditions [46]. TREK-2 phosphorylated by PKC shows low channel activity [40], whereas TRESK dephosphorylated by calcineurin shows high channel activity [41]. These factors may cause different effect of acetaminophen, ibuprofen, nabumetone, and bupropion on TREK-2 and TRESK. The mechanism that modulates these currents will need to be identified to develop specific drugs for depression and pain.
However, at this point, several questions remain to be answered. Are TREK-2 and TRESK inhibitions related to pain? What do TREK-2 and TRESK inhibitions by analgesics used in clinics indicate? DRG and TG neurons predominantly express TREK-2 and TRESK channels among K2P channel family [647]. Single or a combination of TREK-2 and TRESK has been considered as promising therapeutic targets for pain management [12]. The blockade of both TREK-2 and TRESK currents in DRG and TG neurons would cause cell depolarization and increase cell excitability. These agents might result in a reduced effect of pain medication. In this study, we could not determine the relationship between blockade of TREK-2 and TRESK by analgesics and pain. The inhibition of TREK-2 and TRESK channels by analgesics should be considered when assessing the various pharmacological effects produced by analgesics. People respond differently to pain medications because each pain condition is unique. The different effects observed among people could result from TREK-2 and TRESK inhibitions. Further, TREK-2 and TRESK expression could be different among individuals. On the other hand, activation of inhibitory interneurons could reduce pain transmission [48]. Depolarization of resting membrane potential by blockade of TREK-2 and TRESK affect interneurons. To our knowledge, however, there is no report about the expression of TREK-2 and TRESK in inhibitory interneurons. So far, there are no specific modulators of TREK-2 and TRESK channel. Few compounds are known to inhibit TREK-2 and TRESK current. Our data add to the number of compounds that inhibit TREK-2 and TRESK currents and provide new tools to identify TREK-2 and TRESK channels expressed in primary cells, such as TG and DRG neurons.
In conclusion, we found that the drugs with anti-pain effects did not activate TREK-2 and TRESK. Our study addresses the pharmacological modulation of TREK-2 and TRESK channels with special respect to the possible clinical application of alleviating or preventing pain. Future studies should take into account the therapeutic effect of TREK-2 and TRESK on pain, and the mechanism by which analgesics inhibit TREK-2 and TRESK currents.
Go to :
 XML Download
XML Download