

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ischemia-reperfusion (IR) injury is a major contributor to acute myocardial infarction associated with coronary artery disease. Potential mediators of IR are involved in oxidative stress, intracellular and mitochondrial Ca2+ overload, and the accumulation of inflammatory cells in the infarcted myocardial tissue [123]. Additionally, inflammatory and fibrotic processes also play major roles in the extension of myocardial infarction. In this context, transforming growth factor beta 1 (TGFβ1) and tumor necrosis factor alpha (TNFα) have been implicated as important factors that are responsible for triggering and mediating these inflammatory and fibrotic responses [4567]. In response to acute myocardial infarction, TNFα is released from macrophages, monocytes, and mast cells to trigger the inflammatory response and further contribute to the development of contractile dysfunction. Furthermore, TGFβ1 plays a causal role in myocardial fibrosis and diastolic dysfunction [8]. Inhibition of TGFβ1 production is essential in therapy to reduce cardiac fibrosis [689]. These two factors may also play a role in the regulation of decorin (Dcn) expression in this context [10111213], as TNFα and TGFβ1 cooperatively or individually downregulate Dcn gene expression to control biological processes [10].
Animal model studies have suggested the beneficial effects of targeting IR with anti-inflammatory and anti-fibrotic treatments [81415], including Dcn pharmacological or gene therapies [141617]. The Dcn molecule is potentially involved in reverse cardiac remodeling via direct inhibition of TGFβ-induced collagen synthesis and anti-fibrotic effects on patients with end-stage heart failure [17]. Dcn expression reportedly increases in myocardial tissue from patients who had undergone implantation of a left ventricular assist device [17]. Also, agerelated downregulation of cardiac Dcn gene expression following myocardial IR was described [18]. Following myocardial infarction, cardiac-targeted administration of Dcn may impact TGFβ expression. In the rat heart, Dcn levels begin to increase at 1 week, reach a peak at 2 weeks [19], and remain elevated until 8 weeks following myocardial infarction [20], suggesting that Dcn may sequester latent TGFβ1 and thereby control TGFβ activity [21]. In addition, post-infarction Dcn gene therapy mitigates cardiac remodeling and dysfunction by altering infarct tissue noncardiomyocyte dynamics and preventing cardiac fibrosis in combination with inhibition of Smad2/3 activation [22]. These findings confirm the previously reported anti-fibrotic effect of Dcn in hearts [17]. Moreover, a decrease in myocardial fibrosis is another important manner in which Dcn may suppress both systolic and diastolic dysfunction [2324].
NecroXs are a series of compounds that have recently been developed by LG Lifescience [25]. NecroX-5 exhibits significant cell and tissue protective potential against various injury factors, including oxidative stress [25], IR injuries [2627], neomycin [28], nitroprusside [29], and gentamicin [30]. Interestingly, NecroX-7 has been reported to reduce hepatic necrosis and inflammation following IR injury [27]. Recently, we found that post-hypoxic treatment with NecroX-5 reduced oxidative stress and Ca2+ overload in rat heart mitochondria following reoxygenation injury [31] and inhibited sodium nitroprusside-induced cardiac cell death [29]. Furthermore, proteomic analysis revealed that NecroX-5 preserved the cellular level of several antiinflammatory proteins in hypoxia/reoxygenation (HR)-treated hearts. As an extension of this previous study, we investigated whether NecroX-5 plays a role in modulating inflammatory and fibrotic responses in an ischemic heart. As expected, our data demonstrate that NecroX-5 exerts anti-inflammatory and antifibrotic effects against HR injuries via modulation of the TNFα/ Dcn/TGFβ1/Smad2 pathway.
Go to : 
METHODS
Ethics statement
Our experiments were conducted in accord with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication No. 85-23, revised 1996). In addition, these experimental protocols were approved by the Institutional Review Board of Animals at the Inje University College of Medicine, and all procedures were performed in agreement with the Institutional Review Board guidelines for the ethical use of animals.
Isolation of hearts
Eight-week-old male Sprague–Dawley rats (weight, 200~250 g each) were anaesthetized with i.p. injection of sodium pentobarbital (100 mg/kg) [32]. The anesthesia was assessed by verification of a lack of nocifensive movement, such as a tail flick reflex. Then, the heart from each animal was quickly mounted and perfused with normal Tyrode's (NT) solution to remove all blood.
Cardiac perfusion
Hearts were perfused with NT solution for 30 min followed by perfusion with ischemic solution [33] for 30 min. In the HR group, the hearts were then reperfused with NT solution for 60 min. In the NecroX-5 group, NecroX-5 (10 µmol/L in NT solution) was administered to the hearts for 60 min during the reperfusion period. Hearts that were perfused with NT for the entire 120 min were considered the control group.
RNA extraction and real-time PCR
Total RNA was extracted with TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Briefly, frozen tissues (50~100 mg) were ground into powder in liquid nitrogen and then suspended in 1 mL TRIZOL Reagent. Then, RNA was precipitated from the aqueous phase with an equal volume of isopropanol. Following washing in 1 mL 75% ethanol, the RNA pellet was then air-dried and re-dissolved in an appropriate volume of RNase-free water. The RNA was quantified using a spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Then, the cDNA was synthesized using Taqman RT reagents (Applied Biosystems, Waltham, MA, USA), according to the manufacturer's recommendations.
The relative mRNA expression of Dcn, TGFβ1, and TNFα were determined in triplicate using SYBR® Premix Ex TaqTM II kit (TaKaRa, Shiga, Japan) with a CFX96 system. This reaction contained cDNA, SYBR green Taq polymerase mixture, and primer pairs (Dcn, forward 5'-caatagcatcaccgttgtgg-3', reverse 3'-ccggacagggttgctataaa-5'; TGFβ1, forward 5'-accggctttcctgcttctca-3', reverse 3'-cgcccgggttatgctggttgt-5'; and TNFα, forward 5'-ctcccaggttctcttcaagg-3', reverse 3'-ctccaaagtagacctgccc-5'). The mRNA expression levels are presented relative to the determined levels of β-tubulin expression in the corresponding samples.
TNFα production in H9C2 cardiomyoblasts
H9C2 cells were maintained in DMEM with 10% fetal bovine serum. The cells were pretreated for 24 h with NecroX-5 (µmol/ L) before stimulation with LPS (10 ng/mL) for 24 h. After this incubation, the cells were detached from the culture dishes with trypsin and then fixed with 80% methanol for 10 min. Cells were then permeabilized with 0.1% PBS-Tween for 20 min. After incubation with 5% BSA in TBS-T to block non-specific protein binding, the cells were incubated with TNFα (Cat. ab9579, Abcam, Cambridge, UK) for 30 min at room temperature. Then, cells were incubated with the second antibody, Alexa Fluor 488® goat anti-mouse IgG (1:500) for 30 min at room temperature. Normal mouse IgG was used as isotype control. Analysis of >10,000 events by flow cytometry (BD Biosciences, Franklin Lakes, NJ, US) was performed [34].
In addition, TNFα production was measured by ELISA using a rat TNF kit (Cat. RAB0480, Sigma, St. Louis, MO, USA). After stimulation of cells with LPS for 24 h in the presence or absence of NecroX-5, TNFα levels in H9C2 cell lysates were determined according to the manufacturer's instructions using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
TGFβ content release
An ELISA (Cat. ab119558, Abcam) was used to assess the levels of TGFβ1 in H9C2 cell culture supernatants. Cells were plated in tissue culture plates at a density of 3×105 cells/well at 75% confluence and then pretreated with NecroX-5 (10 µmol/L) or vehicle for 24 h. LPS (10 ng/mL) was then added to the culture media, and then cells were incubated for 24 h. The media was then collected and analyzed according to the manufacturer's instructions using a SpectraMax M2 microplate reader (Molecular Devices). To block protease activity, media was supplemented with 1% fetal bovine serum.
Protein extraction for LC-MS and peptide count protein quantification
LC-MS and a non-labeled peptide count protein quantification method were used to analyze the proteomes of rat hearts as described previously [3536]. Heart proteins were separated via SDS-PAGE on a 12% polyacrylamide gel. Protein bands were sliced from the gels and dehydrated. These proteins were then eluted for tryptic peptide digestion. LC-MS/MS analysis was performed using a ThermoFinnigan ProteomeX workstation LTQ linear ion trap MS equipped with NSI sources (Thermo Electron, San Jose, CA, USA).
Protein annotation and functional network construction
In order to elucidate the molecular functions and biological processes of the identified proteins, the proteins were further categorized and annotated, and functional networks were constructed. Systematic bioinformatics analysis of the proteomes from the HR- and HR+NecroX-5-treated hearts was conducted using STRING 10 (Search Tool for the Retrieval of Interacting Genes/Proteins) [37].
Western blot
Myocardial tissues and H9C2 cells were homogenized in ice-cold RIPA buffer (25 mmol/L Tris·HCl [pH 7.6], 150 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail, and phosphatase inhibitor cocktail). The homogenate sample was sonicated briefly before centrifugation at 13,500 ×g for 30 min at 4℃. The BCA assay kit (Pierce, Rockford, IL, USA) was used to quantitate protein. Protein (50~100 µg) was separated by SDS-PAGE on a 10~12% polyacrylamide gel. Proteins were then transferred to nitrocellulose membranes (GE Healthcare, Chalfont St. Giles, UK) and blocked in 5% skim milk for 2 h at room temperature. Membranes were then probed with mouse or rabbit polyclonal antibodies to Dcn, Smad2, or pSmad2 (Abcam), or with mouse monoclonal antibodies to α-tubulin (Sigma-Aldrich) at a 1:500 dilution overnight at 4℃. Following three washes in PBS with Tween 20 (PBS-T, pH 7.4), membranes were incubated with horseradish peroxidase-conjugated antimouse or anti-rabbit IgG secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) at a 1:2000 dilution for 2 h at room temperature. Following washing with PBS-T three times, a western blotting detection kit (Abclon Inc., Seoul, Korea) was utilized to visualize the antibody-bound proteins. These experiments were performed in triplicate.
Statistical analysis
Data are presented as means±standard error of the mean (SEM). One-way analysis of variance (ANOVA) was used to assess the differences between the control and the treatment. In addition, a control-to-treatment comparison with respect to time was performed using two-way ANOVA, using Origin 8.0 software (OriginLab, Northampton, MA, USA). p-values of ≤0.05 were considered statistically significant.
Go to :
RESULTS
Alterations of Dcn expression were assessed using LC-MS and real-time PCR
Protein expression profiling of NecroX-5-treated HR, untreated HR, and control hearts was performed using LC-MS. The Dcn protein expression levels in hearts experiencing HR and NecroX-5-treated hearts are shown in Fig. 1A (red bar). While HR significantly reduced Dcn protein levels, post-hypoxic treatment with NecroX-5 strongly dampened this reduction. Dcn protein levels in control, HR, and NecroX-5-treated hearts were 110.1±14.2%, 68.8±6.3%, and 106.0±4.5%, respectively. Consistent with the proteomic findings, the mRNA expression levels of Dcn were also markedly higher in NecroX-5-treated hearts (77.05±0.72% compared to control) than those in untreated HR hearts (45.91±10.05% compared to control; Fig. 1A, black bar).
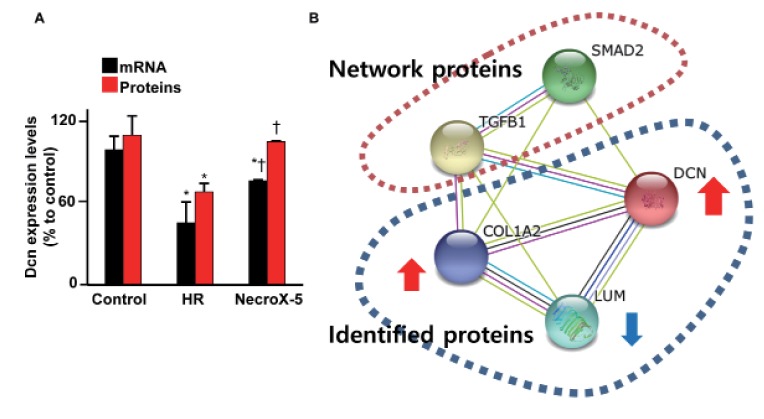 | Fig. 1Proteomics and real-time PCR analysis for Dcn expression levels in cardiac tissue.(A) Dcn mRNA (black bar) and protein (red bar) expression in control, HR-, and NecroX-5-treated post-HR hearts were examined using quantitative real-time PCR and proteomics analysis. n=3~4/group; *p<0.05 vs. control, †p<0.05 vs. HR. (B) Network analysis of identified proteins (DCN, COL1A2, and LUM) and predicted networking proteins (TGFβ1 and SMAD2). Network analysis was performed using STING v10 [37].
|
Alterations of Dcn and TGFβ1/Smad2 reveal antiinflammatory and anti-fibrotic properties of NecroX-5
Our previous proteomics study and network analysis [35] revealed a small protein network that comprised three identified proteins (Dcn, collagen alpha-2[I] chain [Col1a2], and lumican [Lum]) and two predicted networking proteins (TGFβ1 and SMAD2; Fig. 1B). These identified proteins function in the modulation of inflammation and fibrosis [10] and interact with TGFβ1 and SMAD2 [17]. Based on this prior network analysis, we further investigated the role of NecroX-5 in Dcn/TGFβ1/Smad2 alterations in HR-treated hearts and LPS-treated myoblasts.
Dcn has been proposed to be a physiological inhibitor of TGFβ1, suggesting that alterations of Dcn may be related to expression of TGFβ1 and the downstream factor Smad2. We examined TGFβ1 expression in order to test whether NecroX-5 attenuates inflammatory and fibrotic processes (Fig. 2). In agreement with our proteomics and real-time PCR results, we observed a significant increase in Dcn protein levels in NecroX-5-treated hearts compared to HR-treated hearts by western blot (1.06±0.01 and 0.69±0.06, respectively; p<0.05; Fig. 2B), while Dcn expression in control hearts was 1.11±0.14 (ratio to control). In addition, we examined the protein expression levels of Smad2 and pSmad2 in total cardiac protein samples. The highest ratio of pSmad2/Smad2 protein was found in HR-treated rat hearts (1.38±0.01, Fig. 2A, 2C). Interestingly, that ratio was decreased in NecroX-5-treated hearts. In control hearts and post-hypoxic treated NecroX-5 hearts, pSmad2/Smad2 ratios were 1.10±0.1 and 1.15±0.01, respectively (Fig. 2A, 2C). Furthermore, mRNA TGFβ1 expression levels were significantly higher in HR-treated hearts and NecroX-5-treated HR hearts (4.31±1.46 and 1.00±0.19, respectively; p<0.05) than in control hearts (0.54±0.05; Fig. 2D); however, these elevated TGFβ1 cytokine levels were markedly attenuated in NecroX-5-treated HR hearts compared to those in the HR-treated hearts.
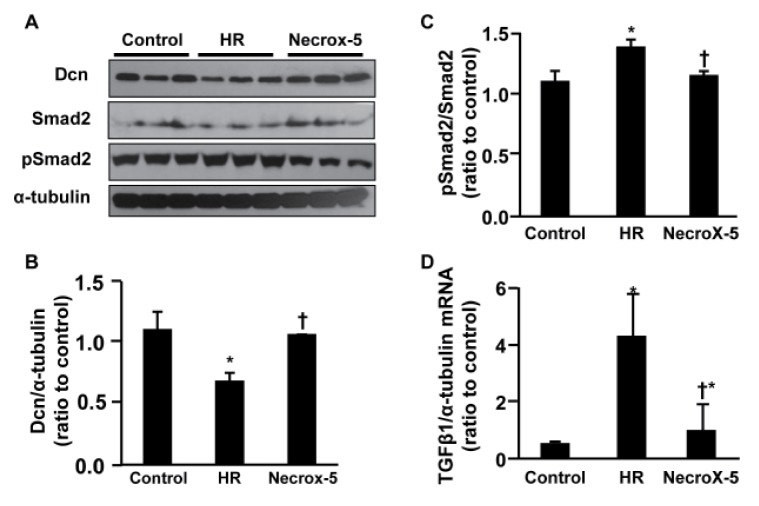 | Fig. 2Effect of NecroX-5 on the expression levels of Dcn, Smad, and TGFβ1 in HR-treated cardiac tissue.Immunoblotting was performed for Smad2, pSmad2, Dcn, and α-tubulin (A). Quantitation of the Dcn/α-tubulin ratio (B) and pSmad2/Smad2 ratio (C) are shown. Real-time PCR was performed to assess the mRNA expression of TGFβ1 relative to α-tubulin expression in hearts (D). n=3~4 for each group; *p<0.05 vs. control, †p<0.05 vs. HR.
|
To further investigate the anti-inflammatory and anti-fibrotic effects of NecroX-5, we measured the levels of TGFβ1, Smad2, and pSmad2 in inflammatory H9C2 cultured cells. As LPS is widely used to stimulate the inflammatory response [34], we pretreated the cultured H9C2 cells with NecroX-5 (10 µmol/L) for 24 h prior to treatment with LPS for 24 h. Inflammation was confirmed by the increased TNFα expression levels in these cells following LPS stimulation (Fig. 3A~C). Supplementation with either NecroX-5 or LPS significantly stimulated TNFα mRNA (Fig. 3A) and protein (Fig. 3B) levels compared to treatment with vehicle alone; however, NecroX-5 treatment effectively suppressed the LPS-stimulated transcription of this cytokine. More specifically, TNFα production was markedly increased to 3.02±0.06 ng/ml (Fig. 3B). Pretreatment with NecroX-5 reduced the observed increases in TNFα production to 1.95±0.05 ng/ml. No significant difference between TNFα levels in the cells treated with NecroX-5 (2.16±0.04 ng/ml), and those receiving control treatment (2.13±0.03 ng/ml) was detected.
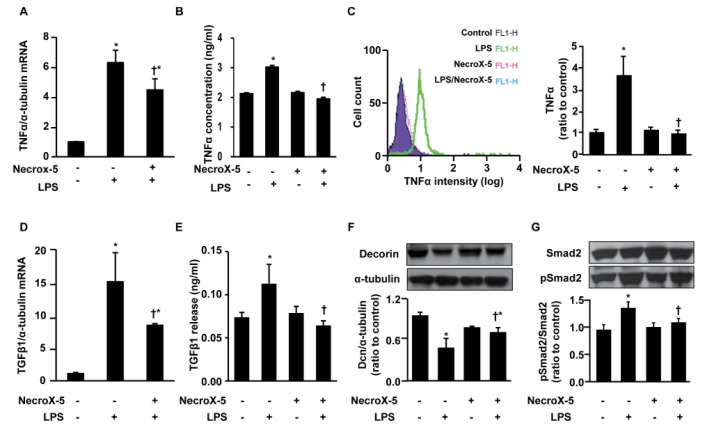 | Fig. 3Effect of NecroX-5 on the expression level of TNFα, TGFβ1, Dcn, and Smad in LPS-treated H9C2 cells.The mRNA expression levels of TNFα relative to α-tubulin were evaluated by real-time PCR (A), and the TNFα protein concentration in cell lysates was evaluated by ELISA (B). FACS analysis yielded the overlay histogram (left) and graph (right) showing the TNFα levels in LPS-stimulated H9C2 cells with or without supplementation of NecroX-5 (C). The mRNA expression levels of TGFβ1 relative to α-tubulin (D) and TGFβ1 secretion (ng/ml) into the culture media (E) were evaluated by real-time PCR and ELISA assay kit, respectively. Western blot analysis was used to determine the expression of Dcn and α-tubulin (F) and Smad2 and pSmad2 (G). H9C2 cells were pretreated with NecroX-5 (10 µmol/L) for 24 h and then LPS (10 ng/ml) was added for 24 h before harvesting. Flow cytometry involved the acquisition of >10,000 events. n=6 for each group; *p<0.05 vs. control, †p<0.05 vs. LPS.
|
Using FACS analysis, we also found that NecroX-5 treatment strongly suppressed TNFα production in LPS-stimulated H9C2 cells (Fig. 3C). As shown in the histogram (Fig. 3C, right panel), TNFα production was highest in the LPS-treated H9C2 cells (3.67±0.86, ratio vs. control), and NecroX-5 supplementation significantly reduced TNFα production to 1.15±0.17 and 0.99±0.15 (ratio vs. control) of the NecroX-5-treated cells and the NecroX-5 and LPS co-treated cells, respectively. Moreover, NecroX-5 strongly suppressed the LPS-stimulated transcription of TGFβ1 in H9C2 cells. The supplementation of either NecroX-5 or LPS significantly stimulated TGFβ1 mRNA expression in comparison to vehicle; however, NecroX-5 effectively suppressed the LPS-stimulated transcription of this cytokine (Fig. 3D). The TGFβ1 mRNA expression level in LPS-stimulated cells was 15.76±4.6 (ratio to α-tubulin), while supplementation by NecroX-5 significantly reduced these levels to 8.81±0.27 (ratio to α-tubulin).
In order to determine whether a decrease in the transcription of TGFβ1 leads to a corresponding reduction in protein secretion, we performed ELISA on media or lysates from H9C2 cell cultures stimulated with the inflammatory protocols described above. As expected, the released TGFβ1 concentrations were significantly diminished in samples treated with NecroX-5 and LPS compared to those treated with LPS alone (Fig. 3E). After 24 h of LPS stimulation, TGFβ1 protein levels in control, LPS, NecroX-5, and NecroX-5 plus LPS cultures were 0.073±0.007, 0.112±0.023, 0.076±0.008, and 0.068±0.009 ng/ml, respectively (Fig. 3E); however, no significant difference between the NecroX-5-treated cells and the LPS and NecroX-5 co-treated cells was detected. Overall, these data demonstrate that treatment of LPS-stimulated H9C2 cells with NecroX-5 strongly suppresses TGFβ1 production.
Furthermore, following LPS stimulation, levels of Dcn, Smad2, and pSmad2 in H9C2 cells were also examined by western blotting analysis (Fig. 3F, 3G). LPS treatment significantly reduced Dcn protein levels in H9C2 cells (Fig. 3F). The ratio of Dcn expression to α-tubulin expression in LPS-stimulated H9C2 cells was 0.48±0.14, while in the control group, this value was 0.94±0.03 (ratio to control). NecroX-5 treatment did not increase Dcn protein level compared to control; however such treatment attenuated the LPS-induced reduction in Dcn expression (0.69±0.07, ratio to control). In addition, LPS treatment significantly increased phosphorylation of Smad2 (pSmad2/ Smad2 ratio), a downstream target of TGFβ1 in the cellular inflammation response (1.33±0.12, ratio to control; Fig. 3G). This increase was strongly attenuated in cells treated with NecroX-5 (1.05±0.01, ratio to control), although NecroX-5 treatment did not affect the phosphorylation level of Smad2 (0.99±0.01, ratio to control). Collectively, these data suggest that in the presence of pro-inflammatory stimulation, NecroX-5 suppresses the activation of TNFα, an upstream inhibitory regulator of Dcn, which further suppresses TGFβ1/Smad2-mediated inflammation.
Go to :
DISCUSSION
In this study, we described the novel action of NecroX-5 as an anti-inflammatory or anti-fibrotic agent in acute HR injury or chronic inflammation via modulation of the TNFα/ Dcn/TGFβ/Smad2 signaling pathway (summarized in Fig. 4). Fibrosis and inflammation are major pathological responses and represent a hallmark of cardiac remodeling associated with impaired contractility and reduced cardiac function [173839]. To date, multiple experimental studies demonstrated a dramatic reduction in cardiac malfunction following use of specific antiinflammatory and anti-fibrosis strategies, such as modulation of TNFα and TGFβ expression [1440]. Importantly, both TNFα and TGFβ also potentially contribute to the regulation of Dcn expression [10111213].
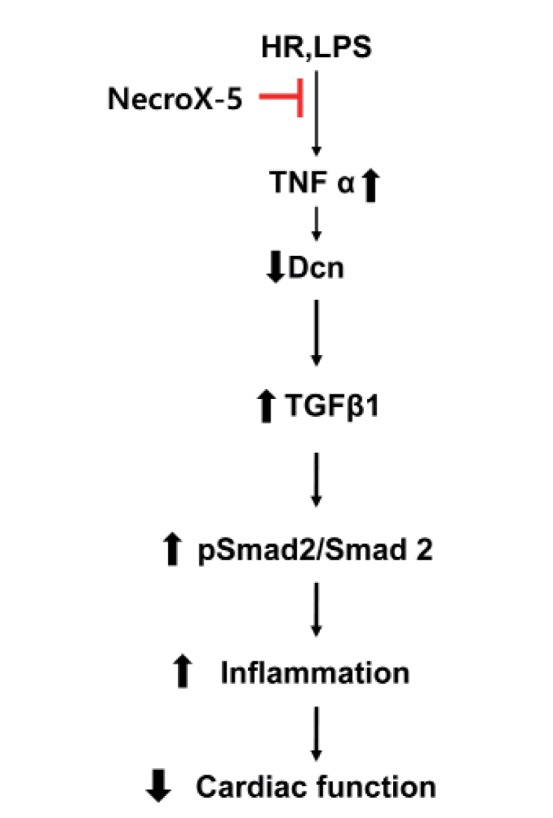
Given the fact that TGFβ plays an important role during healing after myocardial infarction by inducing fibroblast proliferation, increasing extracellular matrix protein synthesis, driving scar formation, and subsequently affecting cardiac remodeling and function [9224142], considerable attention has focused on development of new compounds to suppress TGFβ activity [8]. Accordingly, blockade of the TGFβ/Smad pathway reduced fibrosis and improved cardiac function [843]. Among the most recent successful interventions, Dcn has been suggested to be a physiological inhibitor of TGFβ and to limit the duration of the TGFβ response during inflammation, fibrosis, and tissue repair [1744]. The ability of Dcn to prevent tissue fibrosis and promote tissue regeneration has been described for various injury models [1617444546]. Dcn has been reported to function in reverse cardiac remodeling via direct inhibition of TGFβ-induced collagen synthesis [17]. The function and tissue expression of Dcn has led to the suggestion that this factor plays a protective role following myocardial infarction [1847]. In rat hearts, Dcn levels begin to increase and remain elevated for weeks, suggesting that Dcn may sequester latent TGFβ1 and thereby exert beneficial control over TGFβ activity [192021]. In addition, recent evidence from recombinant adeno-associated viralmediated Dcn overexpression suggests that Dcn exerts potential anti-fibrotic effects to inhibit cardiac fibrosis [48]. In agreement with a previous report [17], post-infarction Dcn gene mitigates the adverse effects on left ventricular geometry and function by affecting cardiac fibrosis and infarct tissue dynamics through the TGFβ/Smad2/Smad3 pathway. Myocardial fibrosis contributes to both systolic and diastolic dysfunction [2324], and thus, reduction of this fibrosis is likely another important way that Dcn may modulate left ventricular remodeling and improve cardiac function. Moreover, Dcn gene therapy also significantly increases proliferation of both cardiac myofibroblasts and vascular endothelial cells, suggesting this gene modulation preserves the infarct wall thickness, reduces wall stress, and mitigates left ventricular dilatation and dysfunction [22]. Exercise has a various benefit for the cardiac protection by itself or combined with other medicines [4950]. Interestingly, chronic exercise is another stimulating factor of Dcn expression and release from skeletal muscle [17]. These findings could suggest the Dcn can be a mediator of exercise-induced anti-inflammation or anti-fibrosis response.
In our study, NecroX-5 treatment induced markedly higher Dcn expression levels than those observed in untreated hearts (Fig. 2). We found that this increase in Dcn was associated with a concomitant decrease in TGFβ1 expression. Thus, the roles of NecroX-5 on the downregulation of TGFβ1 via Dcn may be explained by two possibilities. Firstly, NecroX-5 may inhibit transcription and translation of TGFβ1, subsequently eliminating the inhibitory effect of this cytokine on the Dcn gene expression (i.e., a downregulated regulator of Dcn). Secondly, lower TGFβ1 in NecroX-5-treated hearts may result from direct binding of Dcn to TGFβ1 with high affinity [51]. Consistent with a previous report [52], increased Dcn production may indicate a mechanism by which cells counteract an injury induced via high glucosestimulated TGFβ1. This signaling pathway was also reported by Kahari et al. following treatment with dexamethasone [51]. Nevertheless, conflicting data also demonstrate that the modulation of TGFβ1 via Dcn expression is altered in different cell types [101113], leading to questions of whether the Dcn-TGFβ1 complex inactivates or enhances TGFβ activity [53].
Smad2 protein is generally considered a downstream mediator of TGFβ signaling in myocardial inflammation and fibrosis. TGFβ1-stimulated cardiomyocytes exhibit considerable phosphorylated Smad2 (pSmad2) [22535455]. In agreement with previous studies [843], the lower pSmad2 production observed in NecroX-5-treated samples (Fig. 2, 3) may contribute to a reduction in fibrosis and inflammation, supporting the possibility of cardiac function preservation by NecroX-5. Conversely, earlier studies reported that blockade of the TGFβ/Smad pathway reduced fibrosis and improved cardiac function [5354], perhaps via regulation of calcium homeostasis [31]. Thus, downregulation of the TGFβ1/Smad2 pathway by NecroX-5 could potently improve cardiomyocyte Ca2+ handling and further complement the effect on calcium homeostasis as described in our previous report [6]. Activation of TGFβ/Smad signaling, however, could reduce or increase cardiac function [56]. Previous studies suggest that Dcn is required for the TGFβ response without changes in the Smad-dependent pathway in myoblasts [12]. Furthermore, increased sequestration of Dcn with TGFβ would also promote cell proliferation [734].
Additionally, TNFα is another important mediator of cardiac inflammation [10] and a potent downregulator of Dcn gene expression. In particular, this cytokine exerts its maximal inhibitory effect on promoter sequences relative to the transcription start site of Dcn gene [13]. In our study, TNFα mRNA and protein levels were dramatically increased in LPS-stimulated inflammatory H9C2 cells. Interestingly, these increases were significantly diminished in the presence of NecroX-5 treatment. Consisted with a previous report [10], we found that in inflamed H9C2 cells, TNFα upregulation was accompanied by Dcn downregulation. These alterations led to increased TGFβ1 expression and subsequently resulted in increased Smad2 phosphorylation and elevated inflammation. In this context, TNFα may act independently, as well as exert an additive inhibitory effect with TGFβ1, on Dcn expression. Thus, NecroX-5 administration may beneficially control biological processes via cooperative or specific modulation of TNFα, Dcn, TGFβ1, and Smad2 expression [5758].
Moreover, inflammation-mediated mitochondrial alterations play a critical role in inflammation-linked diseases [4259]. Limiting inflammation and consequent mitochondrial function modulation may offer promising therapeutic targets for treatment of ischemic disease. As a result, pharmacological treatments that target mitochondrial pathways may be pursued as a means to treat inflammatory responses [60]. Previously, TNFα was found to modulate inflammation-related mitochondrial function via effects on the mitochondria [60]. These effects included alterations in the mitochondrial complex activity, loss of mitochondrial membrane potential, and decreases in energy production. Mitochondrial Ca2+ and mitochondrial reactive oxygen species, in turn, are primary mediators of TNFα-mediated inflammatory responses [61]. Typically, damaged mitochondria exhibit reduced bioenergetics, favoring cell damage and death [6263]. In addition to its interactions with TGFβ and TNFα, Dcn has multiple molecular targets involved in cell growth, and Dcn may also inhibit the production of inflammatory chemokines [64]. Dcn-induced cell cycle arrest via negative modulation of p21 may reduce fibrosis by suppressing proliferation and differentiation of cardiac fibroblasts [31]. In our previous studies, NecroX-5 reduced oxidative stress and preserved mitochondrial function, as evidenced by peroxisome proliferatoractivated receptor-gamma coactivator-1α (PGC1α) expression, mitochondrial complex activity, mitochondrial oxygen consumption, and inner mitochondrial membrane potential (Δψm) against HR injury [3565]. One possible mechanism of the anti-inflammatory effects of NecroX-5 may be that the antioxidant activity successfully prevents hypoxia-induced nuclear factor kappa B (NFκB) and TNFα activation [55]. In addition, our previous study suggested that NecroX-5 removes mitochondrial ROS during LPS-stimulated inflammation, successfully blocking NLRP3 inflammasome activation, as well as NFκB and TNFα activation, in airway epithelial cells [66]. In addition to these findings, the current study suggested that NecroX-5 may regulate the expression of inflammatory factors, including TNFα and TGFβ1/Smad2, in part via modulation of Dcn.
Several limitations of this study need to be mentioned. First, our experiments focused on the effects of Dcn in TNFα/TGFβ1/ Smad2-mediated cardiac inflammation and fibrosis in the ex vivo heart during a relatively short period of time (>2 h), and therefore, further studies are required to provide a more comprehensive understanding of the anti-inflammatory and anti-fibrotic roles of NecroX-5 in an in vivo model with long-term monitoring. Second, excessive ROS production during HR treatment may lead to damage and degradation of various proteins; thus, the direct effect of oxidative stress or NecroX-5 on the protein levels of Dcn in HR-treated hearts should be addressed in future studies.
Go to :
 XML Download
XML Download