

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Huntington's disease (HD) is a neurodegenerative genetic disease characterized by striatal and cortical atrophy, which leads to uncontrolled movements and cognitive symptoms [1]. HD is caused by mutations of CAG triplets, of which there are more than 35 in the huntingtin protein [2]. Although the understanding of the cellular and molecular pathogenesis of HD has improved, effective therapeutic tools against it have not yet been developed. This is largely because the molecular mechanisms of HD are so complex; therefore, an understanding of the pathogenic processes of HD, including transcription, excitotoxicity, mitochondrial defects, and activation of death-signaling, is essential [3456].
It has recently been proposed that mitochondrial dysfunction is a key factor leading to striatal neurodegeneration in human HD patients, as well as in animal models of HD. For example, metabolic alterations are observed in pre-symptomatic carriers, which suggests that mitochondrial dysfunction is an early cause of HD pathogenesis [78]. In addition, impaired activity of respiratory chain complexes and tricarboxylic acid-cycle enzymes has been observed in the brains of HD patients, and reduced activity of complexes II, III, and IV is reported in the caudate and putamen of patients with advanced HD [9]. Furthermore, administration of mitochondrial complex II inhibitors induces selective striatal neuronal loss combined with behavioral deficits [10]. Mitochondria are key intracellular organs that supply ATP, and since they contain redox enzymes that are involved in the transfer of electrons, mitochondrial dysfunction generates reactive oxygen species. The central nervous system (CNS) has a high demand for energy, and thus mitochondrial dysfunction in the CNS leads to energy failure and results in neurodegenerative disorders, including HD [1112].
Caffeic acid phenethyl ester (CAPE) is a natural compound that has been widely used as a traditional medicine in Asia. CAPE is a polyphenol compound with hydroxyl groups that are believed to be responsible for its biological activities [13]. For example, CAPE induces anti-inflammatory activity by inhibiting arachidonic acid release from the cell membrane and activation of cyclooxygenase–1 and –2 [1415]. Likewise, CAPE suppressed the level of lipopolysaccharide-induced interleukin-1β, interleukin-6, and tumor necrosis factor-α in a macrophage cell line [16]. Furthermore, accumulating evidence has revealed that CAPE has antioxidant, antimicrobial, and anticancer properties [17]. As far as the antioxidant activity of CAPE is concerned, its activity is comparable to that of melatonin in thyroid and liver cells, and it significantly improves the viability of lipopolysaccharide/interferon-γ-treated RAW264.7 cells by significantly reducing the production of nitric oxide and peroxynitrite [1819]. However, although extensive literature is available on the biological activities of CAPE, its therapeutic potential in CNS diseases is not completely understood.
In this study, we investigated whether CAPE could be used as a therapeutic agent against HD. For this purpose, we employed a 3-nitropropionic acid (3NP)-induced striatal neurotoxicity model in mice. As repeated administration of 3NP, a complex II inhibitor, induces histological and behavioral changes reminiscent of the pathogenesis of HD, it is widely used as a chemical model for HD [20].
Go to : 
METHODS
Animals
Male C57BL/6 mice (8 weeks old; Koatech, Kyungki-do, Korea) were housed under standard temperature (22±1℃) and humidity (50%±1%) conditions with a standard 12/12-h light/dark cycle (lights on from 8:00 a.m. to 8:00 p.m.), with access to food and water ad libitum. The animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23, revised 1996), and the experimental protocol was approved by the Institutional Animal Care and Use Committee of the Catholic University of Daegu. Efforts were made to minimize animal suffering and to reduce the number of animals used.
3NP intoxication
The experimental model to induce HD was previously described [1021]. Briefly, 3-nitropropionic acid (3NP; Sigma, St. Louis, MO, USA) was dissolved in saline (25 mg/mL) and passed through a 0.2-µm filter. We followed the 3NP injection protocol described by Huang et al. [21], with 3NP injected intraperitoneally twice daily for two days at 12-h intervals (10:00 a.m. and 10:00 p.m.) at a dose of 60 mg/kg for the first two injections and 80 mg/kg for the second two injections.
CAPE administration
CAPE was dissolved in DMSO and diluted with peanut oil to a final concentration of 2.5 mg/mL in 5% DMSO and 95% peanut oil. The mice in the CAPE-treated group received 30 mg/kg of CAPE for five days (until three days after the last 3NP injection). On the first and second days, CAPE was injected 30 min before the 3NP injection.
Rotarod assessments
The mice were trained on an accelerating rotarod (RotaRod-M; Jeung Do Bio & Plant, Korea) with three trials per day for three consecutive days. Only the mice that attained a steady baseline performance (180 seconds for the fall latency on the second day) were used. The testing was executed from one day before until three days after the last 3NP injection. The mice were tested three times each day at a speed of 4~40 rpm with a 1-h interval between each test. The longest fall latency from the three daily trials was used for the analysis. The data were presented as mean±SEM, and significance was assessed using a log-rank test for the survival results and a two-tailed Student's t-test for the motor behavior results.
Tissue processing
After the last rotarod assessment, the mice were transcardially perfused with cold saline followed by 4% paraformaldehyde in phosphate buffer (0.1 M PB, pH 7.4) under deep anesthesia with 15% chloral hydrate. The brains were post-fixed for 4 h, then cryoprotected in 30% sucrose in 0.1 M PB. According to the Mouse Brain Atlas, sequential coronal sections (40 µm thick) through the striatum (bregma +1.50~–0.50 mm) were prepared using a cryotome (SM2010R; Leica Biosystems, Germany) and every fifth section was processed for Cresyl Violet staining [10].
Cresyl Violet staining and measurement of striatal damage area
For Cresyl Violet staining, the sections were mounted on gelatin-coated slides and dried. After dehydration in a graded alcohol series, the sections were stained for 20 min with 0.1% Cresyl Violet solution (Sigma, St. Louis, MO, USA). After destaining with a solution of 95% ethanol and 0.1% glacial acetic acid, the sections were dehydrated and mounted with Permount (Sigma). To measure striatal neuronal damage, the striatal borders were outlined as described in our previous report [10], and the volumes of the 3NP-induced striatal lesions were measured at every fifth striatal section (200-µm intervals). Photomicrographs of the Cresyl Violet-labeled sections were captured using a digital camera (UC140, VEZU, China) mounted on a microscope. For each section, the total striatal area and the damaged area were outlined, and the areas were measured using Photoshop® software. Volumes were calculated by summing the cross-sectional area of each section, and the percentage of striatal damage in each animal was calculated by dividing the damaged volume by the total striatal volume. Striatal damage was measured on the following scale: no striatal damage, grade 0; less than 10% striatal damage, grade 1; 10–20% striatal damage, grade 2; higher than 20% striatal damage, grade 3. Quantitative measurements of the striatal damage were analyzed with the χ2 test.
Immunohistochemistry
For immunohistochemical labeling, the sections were blocked with 10% normal goat serum in PBS, followed by overnight incubation (4℃) with mouse anti-glial fibrillary acidic protein antibody (1:1,000; Millipore, Temecula, CA, USA) or mouse anti-CD45 antibody (1:1,000; BioLegend, San Diego, CA, USA). After washing, the sections were incubated with secondary antibody conjugated with horseradish peroxidase (1:1,000; Jackson ImmunoResearch, West Grove, PA, USA). After several washes, 0.1% diaminobenzidine (Sigma) and 0.001% hydrogen peroxide were used to visualize the signal.
ABTS assay
To measure the free-radical-scavenging activity of CAPE, the ABTS antioxidant assay was performed according to the manufacturer's instructions (Zenbio, Research Triangle Park, NC, USA). Briefly, 10 µL of CAPE and 20 µL of myoglobin working solution were mixed together. After 100 µL of the ABTS solution was added, the plate was placed on a shaker for 5 min. After the reaction was stopped by adding 50 µL of stop solution, a plate reader was used to measure absorbance at a wavelength of 405 nm. The data were presented as mean±SEM, and significance was assessed using Student's t-test.
Striatal neuronal culture and lactate dehydrogenase (LDH) release assay
The striatal neuronal culture method was described by Lee et al. [22]. Briefly, striatal tissue was dissected from Sprague–Dawley rat pups (E19~E20) and enzymatically digested into a single-cell suspension with a mild protease solution (0.25% trypsin-EDTA; Gibco, Grand Island, NY, USA) at 37℃ for 30 min. After removal of the digestion solution, the tissue was washed three times in dissociation media containing 90 mM of Na2SO4, 30 mM of K2SO4, 16 mM of MgCl2, 0.25 mM of CaCl2, 32 mM of HEPES, and 0.01% phenol red (Sigma, St. Louis, MO, USA), with a pH of 7.7, and transferred to a tissue-culture medium (Dulbecco's modified Eagle medium [DMEM]; GenDEPOT, Barker, TX, USA) containing 10% fetal bovine serum, 2% B27 (Gibco), and 100 U/ml of penicillin/streptomycin. Next, the tissue was triturated into a single-cell suspension via a 5-ml serological pipette and plated at a density of 2.2×105 cells/cm2 on poly-L-lysine-coated 24-well plates.
3NP intoxication and LDH release assay
Cultured striatal neurons were intoxicated with 5 mM of 3NP in culture media for two days. To test its protective effect, 20 or 50 µM of CAPE was also added. Two days later, the LDH release assay was performed as described by Koh and Choi [23]. The value for LDH release from the vehicle-treated condition was normalized to a value of 1. The data were presented as mean±SEM, and significance was assessed using one-way analysis of variance (ANOVA). Post-hoc tests were performed using the Bonferroni correction.
Go to :
RESULTS
Neuroprotective effect of CAPE by radical-scavenging activity
First, we examined the antioxidant activity of CAPE with an ABTS antioxidant test. This assay measures ABTS+ radical formation, so the result identifies direct radical-scavenging activity by electron transfer. For this study, we tested 1, 20, and 50 µM of CAPE, and as shown in Fig. 1A, the radical-scavenging activity was dose-dependently increased, indicating that CAPE has strong radical-scavenging activity. To test the neuroprotective effects, 20 or 50 µM of CAPE was treated with 5 mM of 3NP for two days, and cytotoxicity was measured with the LDH release assay. As shown in Fig. 1B, 50 µM of CAPE successfully protected the cultured striatal neurons. Of note, these cytotoxicity data coincide with the dose-dependent radical-scavenging activity shown in Fig. 1A. In addition, since we co-treated with 3NP and CAPE, it would be more reasonable to interpret this as CAPE protecting the neurons through direct radical-scavenging activity rather than genetic modulation.
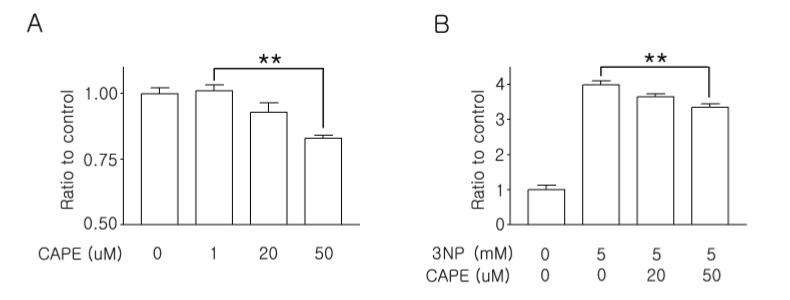 | Fig. 1Neuroprotective effect of CAPE in cultured striatal neurons.(A) The radical-scavenging activity of CAPE was measured with the ABTS antioxidant assay (n=4 for each group). (B) The neuroprotective effect of CAPE against 3NP intoxication was tested. For this study, 3NP and CAPE were treated and an LDH-release assay was performed two days later (n=4 for each group). Data are presented as mean±SEM, and significance was assessed using one-way analysis of variance (ANOVA). Post-hoc tests were performed using the Bonferroni correction. **p<0.01.
|
Neuroprotective effect of CAPE in a chemical model of Huntington's disease
The strong radical-scavenging activity of CAPE and its neuroprotective effect on cultured striatal neurons led us to investigate the therapeutic potential of CAPE in an animal model of HD. For this purpose, a 3NP intoxication model was employed. Repeated administration of 3NP is well known to induce striatal neurotoxicity and behavioral deficits reminiscent of HD [20], so it has been widely used to generate HD in rodents. In this study, CAPE (30 mg/kg i.p.) was administered to mice starting at 30 min before the first 3NP injection and then daily until three days after the last 3NP injection. As shown in Fig. 2A, there was no difference in body-weight change between the two groups. Mortality was 40% in the vehicle-administered group and 11% in the CAPE-administered group (Fig. 2B). Although there was no statistically significant difference between the two groups (log-rank test, p=0.164), these data may suggest that CAPE can ameliorate the overall toxicity of 3NP.
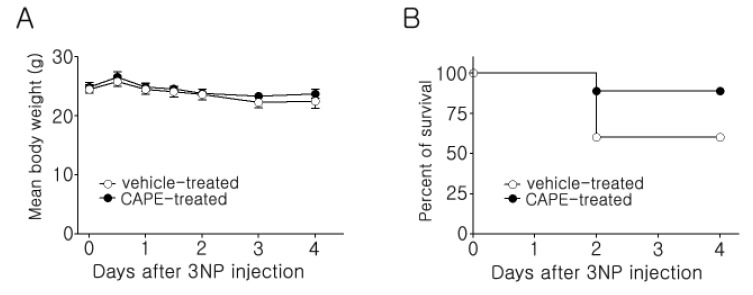 | Fig. 2Body-weight change and survival rates after 3NP injection in mice.(A) Body weight was measured at 12-h intervals during the 3NP injections (60, 60, 80, and 80 mg/kg) and daily for the remaining three days. There was no significant difference between the two groups. Data are presented as mean±SEM, and significance was assessed using Student's t-test. (B) The survival rate was 60% in the vehicle-treated group (6 out of 10 mice survived) and 89% in the CAPE-treated group (8 out of 9 mice survived).
|
3NP induces behavioral deficits reminiscent of HD pathogenesis, so to test whether CAPE rescues behavioral deficits, we employed the rotarod test. This test is used to evaluate motor coordination by measuring the latency to fall from the rotarod [24]. To our surprise, daily administration of CAPE significantly rescued motor deficits on the rotarod. These results suggest the possibility that CAPE may have strong potential as a therapeutic candidate for HD (Fig. 3).
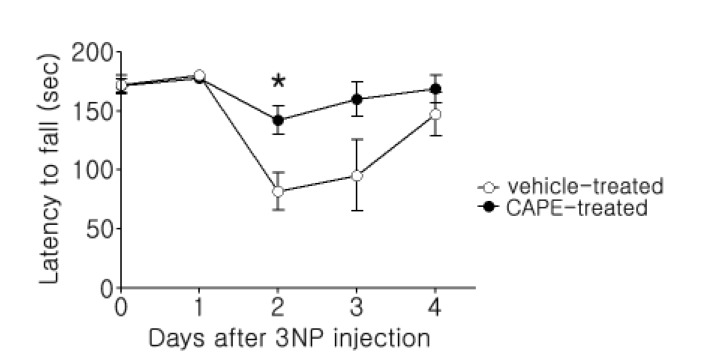 | Fig. 3Rotarod analysis of 3NP-evoked fall latency.Motor activity was tested three times per day on an accelerating rotarod apparatus (4~40 rpm for 3 min), and the longest latency to fall off the rotarod during the three daily trials was used for analysis. The data from the surviving animals until three days after the last 3NP injection were included (n=6 and n=8 for the vehicle- and CAPE-treated groups, respectively). The data are presented as mean±SEM, and significance was assessed using Student's t-test. *p<0.05.
|
Reduced striatal damage after CAPE treatment
Next, we examined striatal neuronal damage with Cresyl Violet staining. The animals were transcardially perfused immediately after the last rotarod behavior test, and the sections were stained with Cresyl Violet. As shown in Fig. 4A, marked striatal damage was observed in the lateral striatum of the 3NP-administered animals. On the other hand, neuronal damage was dramatically reduced by co-administration of CAPE. A statistical analysis of the striatal neuronal damage was performed for the two groups. As shown in Fig. 4B, striatal damage in the CAPE-treated group was significantly reduced compared to the vehicle-treated animals. These data, combined with the behavioral outcomes, clearly suggest strong neuroprotective activity of CAPE against 3NP-induced striatal neurotoxicity.
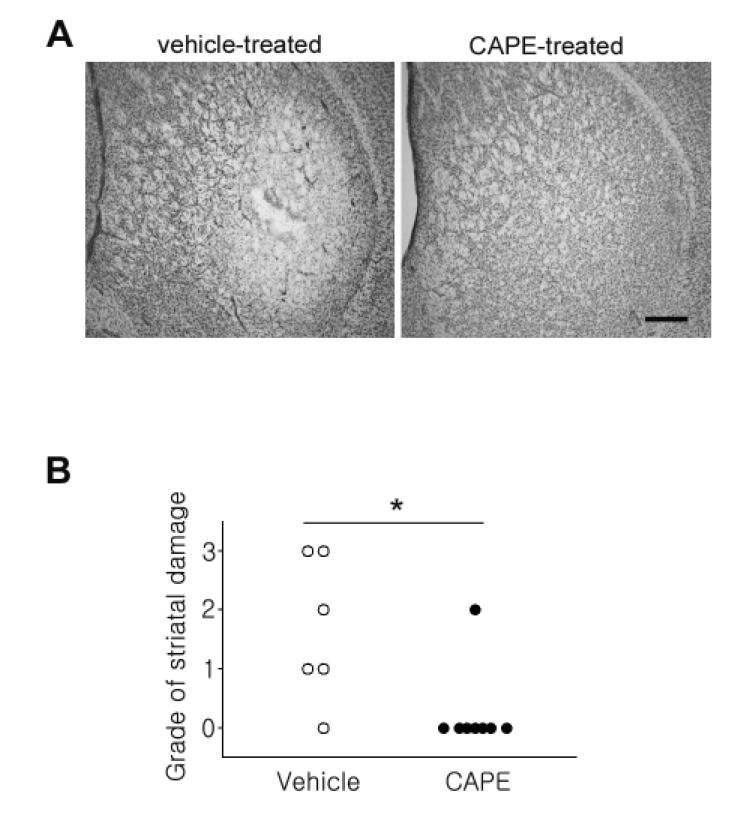 | Fig. 4CAPE reduced 3NP-evoked striatal damage.(A) The mice were perfused three days after the last 3NP injection and striatal damage was assessed with Cresyl Violet staining. The representative image shows distinct striatal damage in a vehicle-treated animal. The scale bar indicates 400 µm. (B) Quantitative analysis of striatal damage in the vehicle- and CAPE-treated groups. Quantitative measurements of striatal damage were analyzed with the χ2 test. *p<0.05.
|
Reduced glial reaction in the CAPE-treated group
We next investigated whether CAPE reduces glial activation in the striatum. Astrocyte activation is a well-known hallmark of 3NP-induced striatal neurotoxicity [25]. For this purpose, immunoreactivity to glial fibrillary acidic protein (GFAP) was examined three days after the last 3NP injection. As shown in Fig. 5, strong immunoreactivity to GFAP was observed in the striatum. However, it was dramatically reduced in the CAPE-treated animals. Microglia are also known to be activated in HD, and these activated microglia mediate inflammatory reactions [2627]. Therefore, we asked whether CAPE also reduces microglia/microphage activation. For this purpose, immunoreactivity to CD45, a marker for activated microglia/macrophages, was investigated. As shown in Fig. 6, this was also dramatically reduced after CAPE treatment.
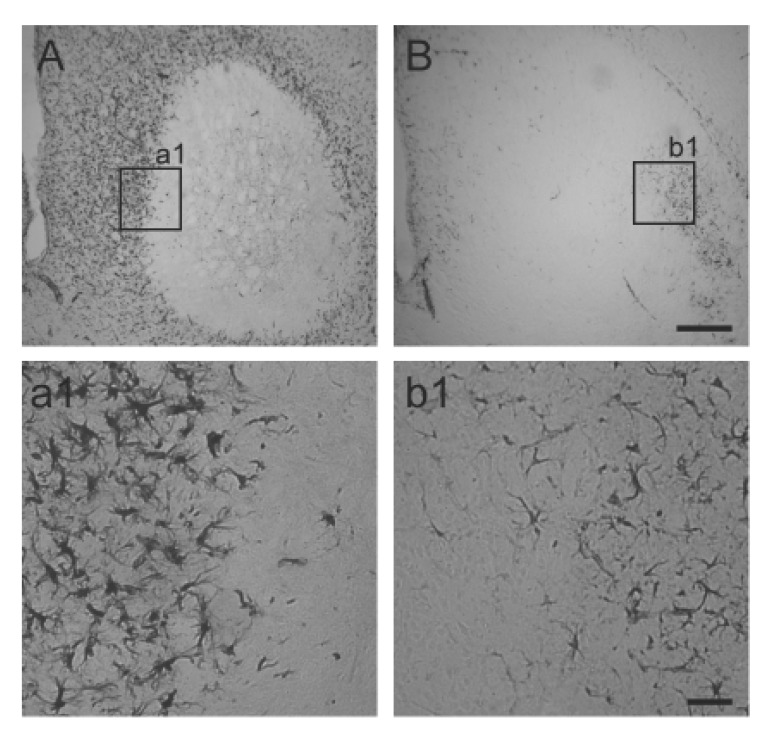 | Fig. 5Astrocyte activation in the striatum.Representative images of GFAP immunoreactivity were shown from vehicle-treated animals (A and a1) and CAPE-treated animals (B and b1). Animals were perfused at 3 days after the last 3NP injection. Size bars are 400 and 50 µm in B and b1, respectively.
|
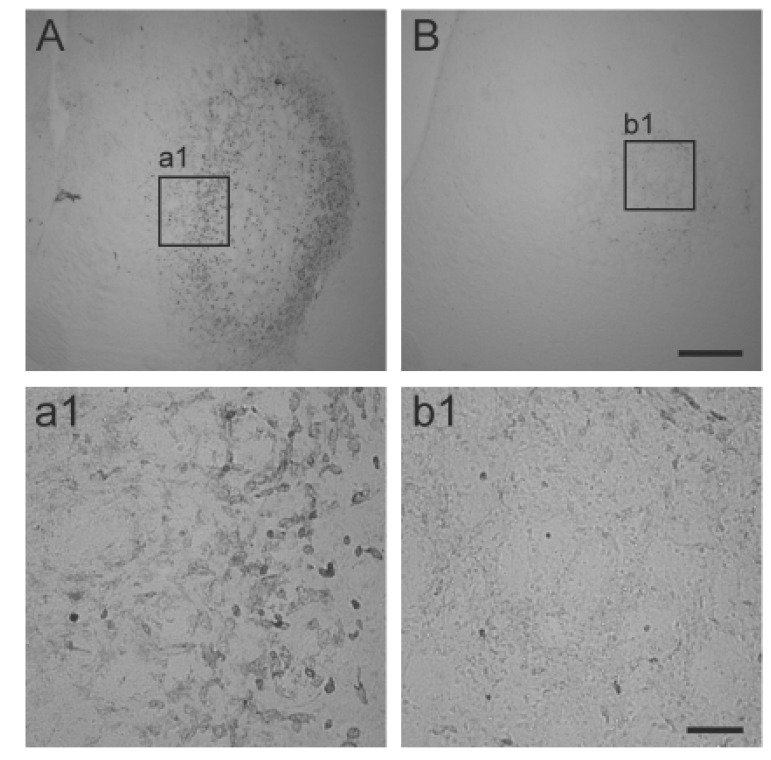 | Fig. 6Microglia/macrophage activation in the striatum.Representative images of immunoreactivity to CD45, a marker of activated microglia/macrophages, were shown from vehicle-treated animals (A and a1) and CAPE-treated animals (B and b1). Animals were perfused at 3 days after the last 3NP injection. Size bars are 400 and 50 µm in B and b1, respectively.
|
Go to :
DISCUSSION
This study was designed to evaluate the therapeutic potential of CAPE in 3NP-induced striatal neurotoxicity in a chemical model of HD. In the in vitro experiment, CAPE showed antioxidant activity, and it significantly reduced the cultured striatal neuronal cell death caused by 3NP. Consistent with these results, CAPE provided neuroprotection and reduced the glial reaction in a chemical model of HD. Furthermore, CAPE significantly ameliorated 3NP-induced behavioral deficits and glial activation. Combined, these observations strongly suggest that CAPE has therapeutic potential against HD.
Many studies have described the diverse biological activities of CAPE, and of note are its potent antioxidant activities. In a sepsis model, CAPE inhibited the cellular level of 3-nitrotyrosine, a marker of peroxynitrite production, and protected cells undergoing lipopolysaccharide/interferon-γ treatment [19]. In adipocytes, CAPE suppressed reactive oxygen species production in a concentration-dependent manner by increasing superoxide dismutase mRNA expression [28]. In the CNS, CAPE reduced acrolein-induced reactive oxygen species generation and glutathione depletion [29]. In addition, CAPE protected dopaminergic neurons from lipopolysaccharide/interferon-γ treatment by inducing the expression of heme oxygenase-1 (HO-1) [30]. Further, CAPE provided neuroprotection against pentylenetetrazol-induced seizures and cigarette smoke by ameliorating oxidative stress [313233]. CAPE was also reported to activate the nuclear factor-erythroid 2 p45 (NF-E2)-related factor 2 (Nrf2) pathway by binding to a cytosolic repressor of Nrf2, Kelch-like ECH-associated protein 1 (Keap1) [3435]. Nrf2 is a master regulator of the gene expression responsible for the antioxidant responses of cells in oxidative environments. It functions as a transcription factor, binding to the antioxidant-response element (ARE) and inducing the expression of a number of antioxidant and detoxification genes, such as NADPH:quinone oxidoreductase 1 (NQO-1) and HO-1 [36]. Along these lines, accumulating evidence indicates that Nrf2 can be a potential target for the treatment of neurodegenerative diseases. For example, activation of the Nrf2-ARE signaling pathway suppressed seizure development and ameliorated cognitive impairment in amygdalakindled rats [37]. In addition, activation of Nrf2 and enhanced expression of Nrf2 downstream antioxidant proteins were responsible for the protective effect of genistein in global cerebral ischemia in rats [38]. Likewise, in N171-82Q mice, a transgenic animal model of HD, it was reported that activation of the Nrf2-ARE signaling pathway rescued motor impairment and striatal atrophy [39]. Combined, these data propose an unequivocal mechanism of CAPE that mediates neuroprotection by genetic modulation of antioxidant proteins.
CAPE is a phenolic compound purified from propolis, and phenolic compounds are known to act as radical-scavengers [4041]. We generated data from an ABTS antioxidant assay showing CAPE's strong radical-scavenging activity. In addition, CAPE provided a protective effect in 3NP-induced neurotoxicity in cultured striatal neurons. It is of interest here that although CAPE has been known to have a strong neuroprotective effect by modulating antioxidant proteins, its direct radical-scavenging activity may also be strong enough to provide protection from 3NP-induced striatal neurotoxicity. To support this idea, we first used 1, 20, and 50 µM of CAPE to measure the radical-scavenging effect, and 20 and 50 µM of CAPE on the LDH release assay. In this experiment, the neuroprotective effect was observed only with 50 µM of CAPE, a result that coincides with the radical-scavenging data. Second, we treated the mice with 3NP and CAPE at the same time. As a strong mitochondrial complex II inhibitor, 3NP rapidly generates reactive oxygen species, including superoxide anions, in the mitochondria [42]. Therefore, although the possibility that expression of antioxidant proteins plays an important role in CAPE-mediated neuronal protection cannot be ruled out, it is postulated that the direct radical-scavenging activity of CAPE is very important in providing neuroprotection. Further research is needed to elucidate the exact neuroprotective mechanism of CAPE in 3NP-induced striatal neurotoxicity.
In cultured striatal neurons, the neuroprotective effect of CAPE was not dramatic compared to in vivo data, with only a 16.2% decrease with 50 µM of CAPE compared to 3NP treatment without CAPE. However, it should be noted that in this study, we selected a very high concentration of 3NP (5 µM); reportedly, maximum radical generation and striatal neuronal cell death is observed at around 1~2 µM of 3NP treatment [4243]. Therefore, the 3NP concentration used in this study was higher than that showing maximum radical generation and toxicity effects, which in turn indicates the strong antioxidant and neuroprotective effects of CAPE.
In summary, we provide for the first time new, solid evidence showing that CAPE has strong radical-scavenging activity and can provide strong neuroprotection against 3NP-induced striatal neurotoxicity under both in vivo and in vitro conditions. Therefore, CAPE has strong potential to provide beneficial effects for HD patients.
Go to :
 XML Download
XML Download