

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Regulatory T (Treg) cells, which express the transcription factor Foxp3, are present in the immune system and are actively engaged in the maintenance of immunological self-tolerance by suppressing self-reactive T cells [12]. They are found at high frequencies in the tumor tissues of various types of cancers, such as breast, lung, liver, pancreatic and gastrointestinal. This subset of T-cells hampers effective anti-tumor immune responses in cancer patients, and these cells can be used as a cellular target to evoke and augment anti-tumor immunity [34].
Cisplatin is commonly used as a chemotherapeutic agent. It clinically displays activity against a wide variety of solid tumors including ovarian, head and neck, and germ cell tumors. However, it does have adverse reactions, such as nephrotoxicity and a drug-resistant phenotype, and these reactions radically limit the clinical utility of cisplatin [56]. Recent studies have shown that the combination therapy of cisplatin with other drugs may be more effective for cancer treatment due to reduced side effects [789].
In a previous study, we reported that the treatment of murine tumors with methyl gallate extracted from Moutan Cortex Radicis exerted antitumor effects. In tumor-bearing animals, treatment with methyl gallate delayed tumor progression and prolonged survival through the inhibition of the tumor infiltration activity of Foxp3+ Treg cells [2]. It was shown that methyl gallate treatment could be used as a potentially useful adjuvant for enhancing the efficacy of anti-cancer therapy by reversing immune suppression. It is envisaged that combinations of methyl gallate, which reduces Treg cells in tumor tissues, with cisplatin, which is antineoplastic drug, may make current cancer therapies more effective. Here, we investigated whether methyl gallate treatment augmented the anti-cancer effect of cisplatin.
METHODS
Material
Methyl gallate was purchased from ChromaDex (purity 96% minimum; Irvine, CA, U.S.) and dissolved in distilled water for 10 mg/ml stock. Stock stored in –20℃ was freshly diluted right before use. Cisplatin was purchased from Sigma-Aldrich (St. Louis, MO, U.S.) and dissolved in normal saline. For Treg depletion, anti-mouse CD25 rat IgG1 (anti-CD25, clone PC61) antibodies were generated in our laboratory from hybridomas obtained from the American Type Culture Collection (Manassas, VA, U.S.). Fluorescence conjugated antibodies for CD3, CD4, CD19, and CD25 detection are purchased from eBioscience (San Diego, CA, U.S.).
Mice
C57BL/6 mice (5~6 weeks of age, weighing 17~20 g) were purchased from Charles River Korea (Seungnam, Seoul, South Korea). Foxp3EGFPC57BL/6 (C.Cg-Foxp3tm2Tch/J) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, U.S.). All mice were kept under specific pathogen-free conditions with air conditioning and a 12 h light/dark cycle. All animal experiments were approved by the Animal Care and Use Committee of Kyung Hee University (KHUASP(SE)-11-033).
EL4 mouse lymphoma cell culture
Mouse EL4 lymphoma (EL4) cells (KCLB 40039; Cancer Research Institute, Seoul, Korea) were cultured in DMEM (Welgene) with 10% FBS (HyClone, Logan, UT) and 1% penicillin-streptomycin (Invitrogen). EL4 cells were treated various doses of methyl gallate (0~50 µg/ml) or cisplatin (0~10 µg/ml). To measure the cell cycle, EL4 cells (2×106 cells/well) were plated in 6-well culture plates and incubated for 24 h in a CO2 incubator. The cells were fixed with 70% ethanol at 4℃ overnight and then incubated with propidium iodide (PI, 0.05 mg/ml) containing RNase (2 mg/ml) for 30 min in the dark. The cells were analyzed by flow cytometry (FACS Caliber, BD Biosciences, U.S.A.). To measure the viability of the cells, EL4 cells (1×105 cells/well) were plated in 24-well culture plates, and MTS ( 20 µl/well) was added to the medium after 24 h.
Immune cell and renal toxicity analysis
Foxp3EGFP mice were injected with saline or methyl gallate (20 mg/kg) once a day for 5 days. Isolated splenocytes were stained with anti-CD4 APC and anti-CD25 PE or anti-CD19 APC and anti-CD3 PE antibodies (eBioscience). After gating CD4+ cells, the CD25+Foxp3+ cells were measured using flow cytometry and the FlowJo software (Treestar, U.S.) for the analysis of Treg cells. To analyze renal toxicity, serum was obtained from each mouse. Blood samples were left at room temperature for 1 hour and then centrifuged at 1,000 g for 10 min. The serum creatinine and BUN levels were measured using a FUJI DRI-CHEM 3500i instrument (Fujifilm Photo LTD., Japan).
Tumor inoculation and drug administration
On day 0, C57BL/6 and Foxp3EGFPC57BL/6 mice were inoculated s.c. in the right flank with 5×104 EL4 cells. To induce the depletion of Treg cells, anti-CD25 Abs (hybridoma clone PC61; 500 µg/mouse) were administered i.p. on days 3, 9, and 15. On day 5 after tumor inoculation, methyl gallate (20 mg/kg, every other day) or cisplatin (1 mg/kg, on days 5, 11, and 17) was injected i.p. The control groups were given saline as a vehicle according to their compared groups. The tumors were measured using two perpendicular diameters every other day, and the volume was then calculated using the following formula:
Confocal microscopy image analysis
To visualize and quantify the degree of migration of the Treg cells into the tumor, lymphoma tissues from Foxp3EGFPC57BL/6 mice were fixed in 4% paraformaldehyde overnight and then in 30% sucrose in PBS for 2 days. Then, the fixed-tumor tissues were embedded in Tissue-Tek O.C.T. compound, frozen, and then sliced into 20-µm sections using a cryostat. The samples were analyzed with a LSM 5 PASCAL confocal laser-scanning microscope system (Carl Zeiss SMT, Berlin, Germany). The numbers of EGFP-positive cells were counted in three lymphoma sections.
RESULTS
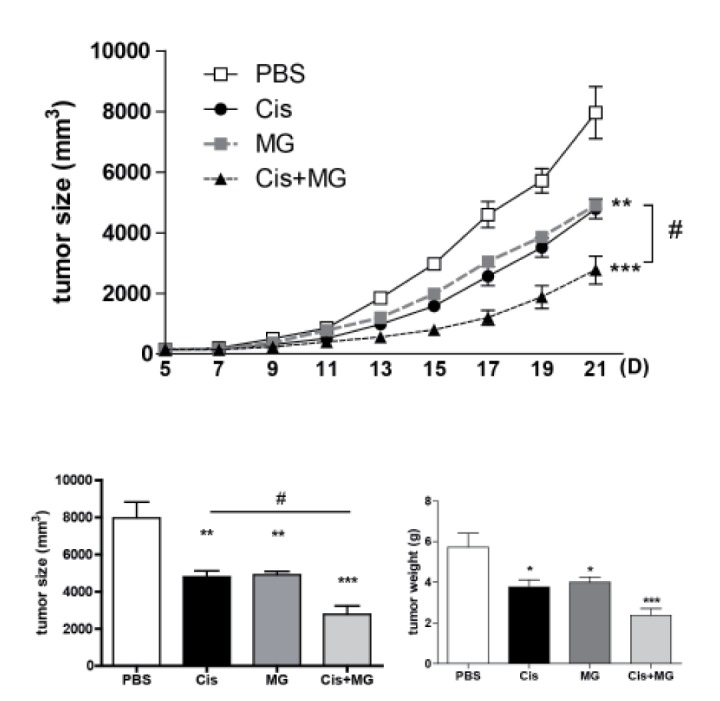
Methyl gallate treatment enhances the anti-cancer effect of cisplatin
To elucidate the effect of the combination therapy of methyl gallate and cisplatin, either or both compounds were used to treat the C57BL/6 mice with established subcutaneous EL-4 lymphoma. Treatment with methyl gallate or cisplatin showed an anti-tumor growth effect. The combination therapy of methyl gallate and cisplatin exhibited a significantly greater effect on anti-tumor growth than methyl gallate or cisplatin as a single treatment (Fig. 1. tumor weight: saline: 5.72±0.69 g, methyl gallate: 3.99±0.25 g, cisplatin: 3.76±0.34 g, methyl gallate+cisplatin: 2.36±0.34 g on day 21). This finding indicates that methyl gallate has an anti-tumor effect and that its mechanism does not hinder the effect of cisplatin.
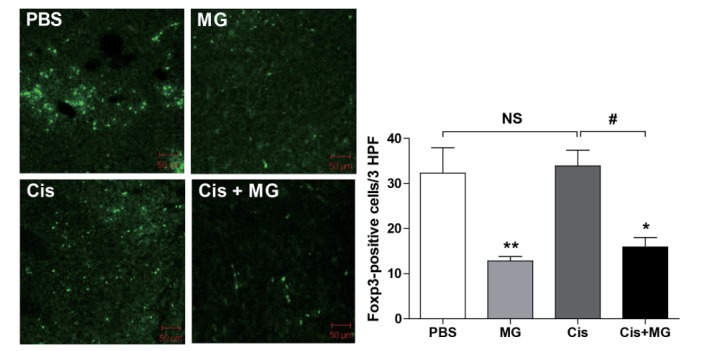
Treg cell infiltration into tumor tissue was inhibited by methyl gallate treatment
To confirm the effect of methyl gallate on Treg cell migration, confocal microscopy was used to inspect cryosectioned tumor tissue from Foxp3EGFP mice sacrificed at day 21 after tumor inoculation. The number of Foxp3-positive cells was significantly decreased in the tumors of the methyl gallate treated mice when compared with the number of positive cells in the tumor tissues of the non-methyl gallate treated controls (Fig. 2. Infiltrated Treg cell number: saline: 32.30±5.61, methyl gallate: 12.80±1.02, cisplatin: 33.88±3.50, methyl gallate+cisplatin: 15.92±2.11). These results indicate that the methyl gallate treatment blocked the migration of Treg cells, which suppressed anti-tumor response in tumor.
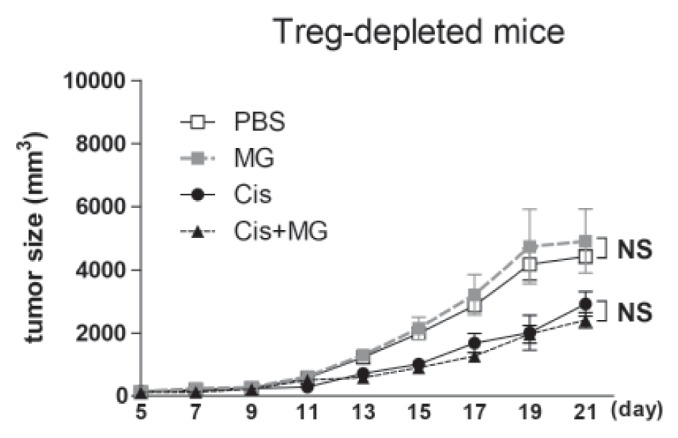
The anti-tumor effect of methyl gallate vanished in Treg cell depleted mice
In a previous report, we found that the anti-tumor activity of methyl gallate was related to Treg cells. To confirm the Treg cell mediated anti-tumor effect of methyl gallate, we treated Treg cell-depleted tumor-bearing mice with methyl gallate or/and cisplatin. The Treg cells were depleted with the anti-CD25 antibody (PC61) in tumor-bearing mice. As expected, in Treg cell-depleted mice, the tumor growth was slower than in wild type mice with vehicle treatment. Cisplatin inhibited the tumor growth of both the Treg cell-depleted and wild type mice, but the methyl gallate treatment lost its anti-tumor effect in the Treg cell-depleted mice (Fig. 3). This result confirms that Treg cells are necessary for the antitumor effect of methyl gallate.
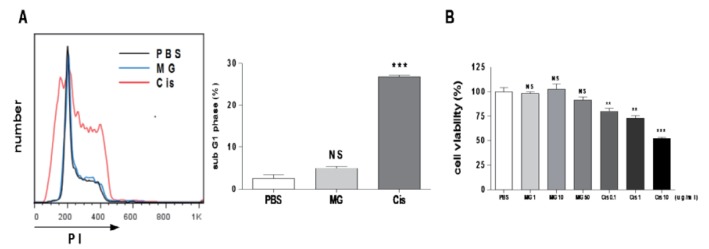
Methyl gallate has no effect on EL4 cell cycle and viability
To determine whether methyl gallate has a direct anti-tumor effect on EL4 cells, we conducted an in vitro tumor cell cycle analysis and a viability test using various concentrations of methyl gallate (0~50 µg/ml). The cisplatin treatment resulted in G1 arrest (% of sub G1 phase: PBS: 2.55±0.90, methyl gallate: 4.98±0.42, cisplatin: 26.86±0.43) and cell death, but the methyl gallate treatment showed no effect on the cell cycle or the viability (Fig. 4). These results suggest that methyl gallate controlled tumor growth indirectly.
Methyl gallate did not effect on population of T cells, B cells, and Treg cells in spleen
To determine whether methyl gallate affected the proportions of immune cells, we measured the T cell, B cell, and Treg cell populations of the spleen in methyl gallate treated mice. As a result, there was no significant change in the ratio of CD3+ T cells, CD19+ B cells, and CD4+Foxp3+ Treg cells compared with the ratio of the vehicle treated mice (Fig. 5).
Methyl gallate does not increase renal toxicity
To probe the nephrotoxicity of methyl gallate, we measured the levels of serum creatinine and BUN in normal mice after methyl gallate injections once a day for 5 days. The results showed that the methyl gallate injections did not alter the serum creatinine and BUN levels in normal mice. Next, we measured the serum creatinine and BUN levels after methyl gallate or/and cisplatin treatment in tumor bearing mice. The treatment with methyl gallate or/and cisplatin did not affect the level of serum creatinine and BUN in our study (Fig. 6).
DISCUSSION
It has become evident that the immune-suppressive elements present in cancer patients are critical impediments to the success of cancer immunotherapy [10111213]. One of the obstacles is that Treg cells, which are physiologically present in the immune system, are actively engaged in the maintenance of immunological self-tolerance by suppressing self-reactive T cells [4]. This T cell subset hampers effective anti-tumor immune responses in cancer patients, and because of this, they are thought to be one of the cellular targets that could be utilized to evoke and augment antitumor immunity. Indeed, Treg cells are found at high frequencies in tumor tissues from various types of cancers, such as breast, lung, liver, pancreatic and gastrointestinal cancers and malignant melanoma [3].
Immunotherapies targeting the depletion or functional alteration of Treg cells have been studied, but the clinical benefits are still unclear. The administration of the cell-depleting anti-CD25 monoclonal antibody resulted in tumor eradication in animal models [1415]. Denileukin diftitox is a recombinant fusion protein product of diphtheria toxin and IL-2 that selectively binds to the IL-2 receptor of cells and, following internalization, inhibits protein synthesis. In a clinical study, denileukin diftitox was used to deplete Treg cells. Denileukin diftitox is the subject of numerous clinical trials, but to date, it fails to realize its clinical promise. Because CD25 is expressed on activated effector T cells, denileukin diftitox may also restrain protective antitumor immune responses. Rasku et al. demonstrated that denileukin diftitox depleted various subsets of T cells, including tumor antigen-specific CD8 T cells [16]. Tremelimumab, the anti-CTLA4 antibody, promotes anti-tumor response. However, Khan et al. reported that these results come from effector T cell activation rather than the modulation of Treg cells [17]. Moreover, a Phase III randomized clinical trial with tremelimumab failed to demonstrate a statistically significant survival advantage of the treatment with tremelimumab over the standard-of-care chemotherapy (temozolomide or dacarbazine) in first-line treatment of patients with metastatic melanoma [18].
A new approach is desirable, as Treg cell depletion leads to an increase in the tumor-mediated effector T cell to Treg cell conversion with a diminution in anti-tumor immune responses [19]. It is agreed that a new approach should selectively modulate Treg cells within the tumor microenvironment, rather than their global depletion to minimize the risk of autoimmune manifestations. Notably, we found that methyl gallate extracted from Moutan Cortex Radicis displays a strong anti-tumor effect through disrupting the tumoral homing of Treg cells [2]. In the line with the thought that immunotherapy can complement anticancer chemotherapy [20212223], here, we studied the effect of the combination therapy of methyl gallate with cisplatin. Indeed, it was found that methyl gallate augmented the anti-cancer effect of cisplatin. In methyl gallate treated mice, tumor infiltration was decreased, while there was no difference in cisplatin treated mice. Methyl gallate treatment did not influence on the global population of T cells, B cells, or Treg cells. It was confirmed that the effect of methyl gallate was Treg cell dependent using a Treg cell depleted model.
In Treg cell tissue trafficking, distinct chemokine receptors and integrin molecules implicated [2425]. CXCR4 mediates Treg cell bone marrow trafficking through bone marrow-derived CXCL12. The lymphoid homing molecules CCR7 and CD62L may facilitate lymphoid homing of Treg cell. CCR2, CCR5, and integrins may mediate Treg cell trafficking into inflammatory tissues. For Treg cell migration into tumor tissue, expression of CCR4 is responsible. CCR4 mediated Treg cell trafficking into human ovarian cancer [26], and Hodgkin lymphoma [27]. Previously, methyl gallate treatment down-regulated CCR4 expression on Treg cells in vitro [2]. Alternatively, there is a report showing CCR5 dependent homing of Treg cell to pancreatic tumor [28]. To elucidate the exact role of methyl gallate on Treg trafficking, further study using chemokine receptor knockout mice is needed.
Some plant polyphenols induce antioxidant and anticancer activities, including cancer cell cycle arrest and apoptosis [29]. Recently, two gallic acid-derived compounds isolated from Casearia sylvestris, isobutyl gallate-3,5-dimethyl ether and methyl gallate-3,5-dimethyl ether, showed anti-tumoral potential [30]. In the current study, we investigated the influences of methyl gallate on the EL4 cell cycle and cell viability. The results showed that methyl gallate had no effect on the cancer cell cycle or cell viability, while cisplatin arrested its cell cycle in the G0/G1 phase and showed strong cytotoxicity in cancer cells. Moreover, methyl gallate did not cause renal toxicity in normal and tumor bearing mice. However, a long-term toxicity study is needed to confirm this result for clinical applications. Additionally, the potentially adverse effect methyl gallate may have on the regulation of Treg cells should be studied.
In tumors, Treg cells not only dampen the anti-tumor process through their interaction with tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) but also promote tumor angiogenesis. Facciabene et al. demonstrated that Treg cell recruitment has a key role in establishing the vascular endothelial growth factor A (VEGFA)-rich tumor microenvironment and increasing tumor angiogenesis, whereas the depletion of Treg cells reduces tumor VEGFA levels and tumor vascularization [31]. Although Treg cells can contribute directly to the excessive production of VEGFA and can support endothelial cell recruitment and expansion, other tolerogenic leukocyte populations, such as MDSCs, also produce VEGFA and support tumor angiogenesis [3233]. MDSCs exert suppressive functions and regulate T cell responses through nitric oxide, reactive oxygen species and TGF-β secretion, and they also promote the induction of Treg cells and favor anti-inflammatory responses [3435]. In the advanced tumor microenvironment, TAMs polarize to type 2 macrophage cells, releasing factors that encourage Th2 differentiation and recruitment [36]. They express a distinct set of cytokines and chemokines, including CCL17, CCL22 and CCL24, favoring the recruitment and development of Treg cells. Further studies probing the effects of methyl gallate on TAMs, MDSCs, and angiogenesis are needed.
In summary, in this study, we provide the first evidence that methyl gallate treatment enhances the antitumor effects of cisplatin therapy, which was mediated through the disruption of the tumoral homing of Treg cells, in a murine tumor model. Therefore, a combination treatment of methyl gallate and cisplatin could be used as an approach to potentiate anti-tumor effects.


 XML Download
XML Download