

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Reactive oxygen intermediates elicit the oxidative decomposition of polyunsaturated fatty acids (i.e., lipid peroxidation) leading to the formation of a complex mixture of aldehydic end-products that includes 4-hydroxy-2-hexenal (HHE) [1]. Indeed, HHE is one of the predominant aldehydes produced by lipid peroxidation [2]. While these aldehydic molecules can exert cytotoxic effects, they can also affect cellular functions via alteration of signal transduction, gene expression, and cellular proliferation. In a previous study, we have demonstrated that HHE-mediated accumulation of reactive oxygen species (ROS) may induce redox-sensitive transcription factor, nuclear factor κB (NF-κB), through activation of ERK and JNK, resulting in cellular apoptosis in HK-2 cells [3].
Mammalian sirtuin 1 (Sirt1) is a member of the highly conserved family of nicotinamide adenine dinucleotide-dependent (NAD+-dependent) protein deacetylases, and is widely expressed in nearly all mammalian organs [45]. Because of its dependency on cellular NAD+ levels, Sirt1 actively responds to redox reactions during cell metabolism [6]. Some of the best-characterized functions of Sirt1, which include promoting increased cellular stress resistance and altering cellular metabolism, are mediated by suppression of NF-κB-dependent inflammatory responses [7].
Resveratrol (RSV) is a natural phytoalexin (3,49,5-trihydroxytrans-stilbene) produced by various plants, including red grapes (Vitis vinifera L.), peanuts (Arachis spp.), berries (Vaccinium spp.), and Polygonum cuspidatum, which exerts multiple beneficial metabolic effects [789]. In addition to scavenging ROS, RSV may provide numerous protective effects against chronic inflammatory diseases through the activation of Sirt1 [8].
The present study was aimed at investigating the effect of RSV on HHE-induced oxidative stress in renal collecting duct cells, and characterizing the signaling mechanisms that govern this process.
Go to : 
METHODS
Cell culture and reagents
Mouse cortical collecting duct cells, M1 (ATCC, Manassas, VA, USA) were cultured. Cells were passaged every 3~4 days in 100-mm dishes containing combined Dulbecco's modified Eagle's medium-F-12 medium (Sigma, St. Louis, MO, USA) supplemented with 5% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin (Sigma). The cells were incubated in a humidified atmosphere of 5% CO2 and 95% air at 37℃ for 24 hr, and sub-cultured at 70~80% confluence. For experimental use, M1 cells were plated onto 60-mm dishes in medium containing 5% fetal bovine serum for 24 h and cells were then switched to Dulbecco's modified Eagle's medium-F12 without serum for 16 hr. The cells were harvested at the end of treatment for further analysis. HHE was obtained from Cayman Chemical, Inc. (Ann Arbor, Michigan, USA). RSV (25 µM) and N-acetyl-l-cysteine (NAC, 10 mM) were obtained from Sigma-Aldrich. Bay 11-7082 (10 µM) was obtained from BioMol (Plymouth Meeting, PA, USA).
Nuclear extracts preparation
For nuclear extracts, cells were lysed using NE-PER® nuclear extraction reagent (NER) (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer's protocol. Briefly, M-1 cells incubated with HHE were harvested by scraping into cold PBS, pH 7.2 and then centrifuged at 14,000 × g for 2 min. After removing the supernatant, 100 µL of ice-cold cytoplasmic extraction reagent (CER) I was added to the dried cell pellets. After incubated on ice for 10 min, ice-cold CER II was added to the tube. The tube was centrifuged at 16,000 × g for 5 min and pellet fraction was suspended in 50 µL of ice-cold NER. After centrifuging the tube at 16,000 × g for 10 min, the supernatant (nuclear extract) fraction was transferred to a clean tube [101112].
Western blot analysis
The cells were harvested, washed twice with ice-cold PBS, and resuspended in lysis buffer (20 mM Tris–HCl, pH 7.4, 0.01 mM EDTA, 150 mM NaCl, 1 mM PMSF, 1 µg/ml leupeptin, 1 mM Na3VO4) and sonicated briefly. After centrifugation, the supernatant was prepared as protein extract, and protein concentrations were measured (Pierce BCA protein assay reagent kit, Pierce, Rockford, IL). Equal amounts of protein were separated on 8 or 12% SDS-polyacrylamide gels. The proteins were electrophoretically transferred onto nitrocellulose membranes using Bio-Rad Mini Protean II apparatus (Bio-Rad, Hercules, CA, USA). The blots were blocked with 5% milk in PBS-T (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaCl, and 0.1% Tween-20 at pH 7.5) for 1 hr. The anti-Sirt-1, anti-NOX4, and anti-p47phox (Santa Cruz Biotechnology, Santa Cruz, CA), anti-COX-2 (Cayman Chemical, Ann Arbor, Michigan, USA), anti-extracellular signal-regulated kinases (ERK), anti-phosphorylated ERK (p-ERK), anti-nuclear factor erythroid 2-related factor 2 (Nrf2), anti-Kelch ECH associating protein 1 (Keap1), anti-c-Jun N-terminal kinase (JNK), anti-phosphorylated JNK (p-JNK), anti-phosphospecific P38 MAPK (p-P38 MAPK), and NF-κB p65 (Cell Signaling Technology, Beverly, MA, USA), iNOS (BD Transduction Laboratories, San Joes, CA, USA), anti-IκBα (Santa Cruz Biotechnology, Santa Cruz, CA), Histone H3 (Cell Signaling Technology) and β-actin (Sigma) antibodies were diluted in a blocking buffer and incubated with the blots overnight at 4℃. The bound antibodies were detected with a 1:1000 dilution of horseradish peroxidase-conjugated secondary antibody according to the instructions provided with the ECL kit (Snta Cruz Biotechnology).
Intracellular level of ROS
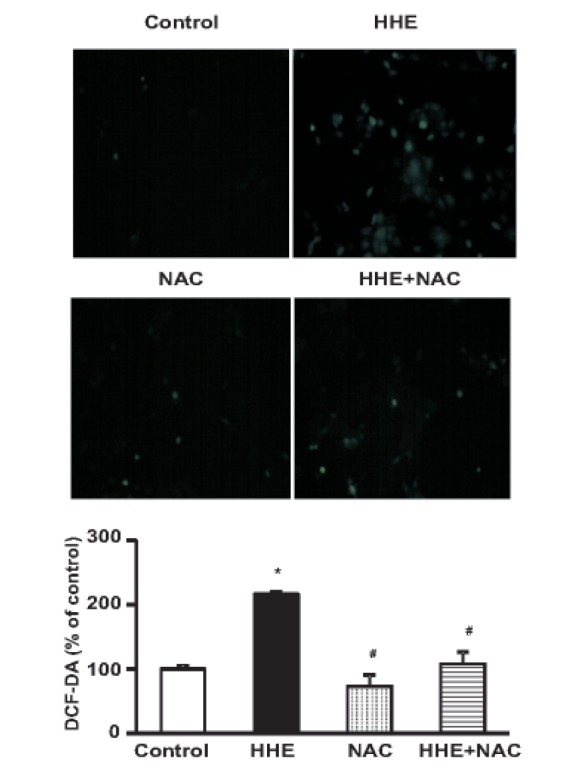
M1 cells were cultured in 24-well plates until they reached confluence. Cells were 1 h pre-incubated with NAC (10 mM) and then treated with 10 µM of HHE for 8 h. At the end of the experimental periods, cells were preloaded with 10 µM 2', 7'-dichlorofluorescein diacetate (DCF-DA; Molecular Probes) for 30 min at 37℃. Fluorescence intensity was analyzed by a fluorescence reader (Fluoroscan Ascent FL; Lab systems, Helsinki, Finland) using 485 nm excitation and 538 nm emission filter. M1 cells were cultured on a 6-well plate for DCF-DA staining. M1 cells were 1 h pre-incubated with NAC (10 mM) and then treated with 10 µM of HHE for 8 h. Cells were washed twice with hanks balanced salt solution (HBSS) and incubated with HBSS (without phenol red) containing DCF-DA for 30 min at 37℃ in dark. The images were obtained with a fluorescence microscope (Nikon, Tokyo, Japan).
Statistical analysis
Results are presented as means±SEM of three individual experiments. Differences were analyzed by ANOVA with post-hoc comparison. Statistical significance of differences was accepted at the level of p<0.05.
Go to :
RESULTS
HHE treatment inhibits Sirt-1 and AQP expression in M1-cells
There was a statistically significant decrease in Sirt1 and AQP2 protein expression in M1 cells after incubation with HHE (10 µM) for 3, 6, and 8 hr (Fig. 1). In addition, there was a trend towards decreased Sirt1 and AQP2 expression after the 8 hr HHE treatment; however, this difference was not statistically significant.
 | Fig. 1Effects of 4-hydroxy-2-hexenal (HHE) on the protein expression of Sirt1 and aquaporin 2 (AQP2) in mouse cortical collecting duct cells (M1 cells).Cells were treated with HHE (10 µM). After 1, 3, 6, and 8 hr, the protein expression of Sirt1 and AQP2 was decreased. Results are presented as means±SEM of three individual experiments. *p<0.05 vs. control.
|
HHE promotes the expression of NADPH oxidase and oxidative stress markers
M1 cells treated with HHE (10 µM) for 1, 3, 6, and 8 hr exhibited increased protein expression levels of NOX4, p47phox, inducible nitric oxide synthase (iNOS), and COX2 (Fig. 2A). Meanwhile, pretreatment of M1 cells for 1 hr with NAC (10 mM) attenuated the effects of HHE on the expression levels of Sirt1, NOX4, COX2, and AQP2 (Fig. 2B). In addition, the formation of ROS was detected using the ROS-sensitive fluorescent DCF-DA in M1 cells. HHE treatment resulted in a dose-dependent increase of DCF fluorescence after incubation of 24 h (data not shown). Fig. 3 shows that treatment of 10 µM of HHE for 8 h increased DCF - DA fluorescent in M1 cells, which was attenuated by 1 h pre-treated 10 mM NAC (Fig. 3).
 | Fig. 2Effects of HHE on the protein expression of NOX4, p47phox, iNOS and COX2.M1 cells were treated with HHE (10 µM). After 1, 3, 6, and 8 hr, protein expression of NOX4, p47phox, iNOS and COX2 were increased (A). Effects of N-acetyl-l-cysteine (NAC) on expression of NF-κB p65 subunit, Sirt 1, NOX4, COX2 and AQP2. The protein expression of Sirt1, NOX4, COX2 and AQP2 was attenuated by 1 hr pre-treated NAC (10 mM) (B). Results are presented as means±SEM of three individual experiments. *p<0.05 vs. control. #p<0.05 vs. HHE.
|
HHE treatment promotes nuclear localization of NF-κB
Fig. 4A depicts the changes in the nuclear localization of the NF-κB p65 subunit in M1 cells treated with HHE (10 µM). There was a concurrent increase and decrease in the nuclear and cytoplasmic protein levels of the p65 subunit and IκBα, respectively, after 30 min of HHE treatment. However, while the nuclear levels of p65 remained steady throughout the time course, there was a steady decrease in the cytoplasmic levels of IκBα during the 2 hr experiment (Fig. 4A).
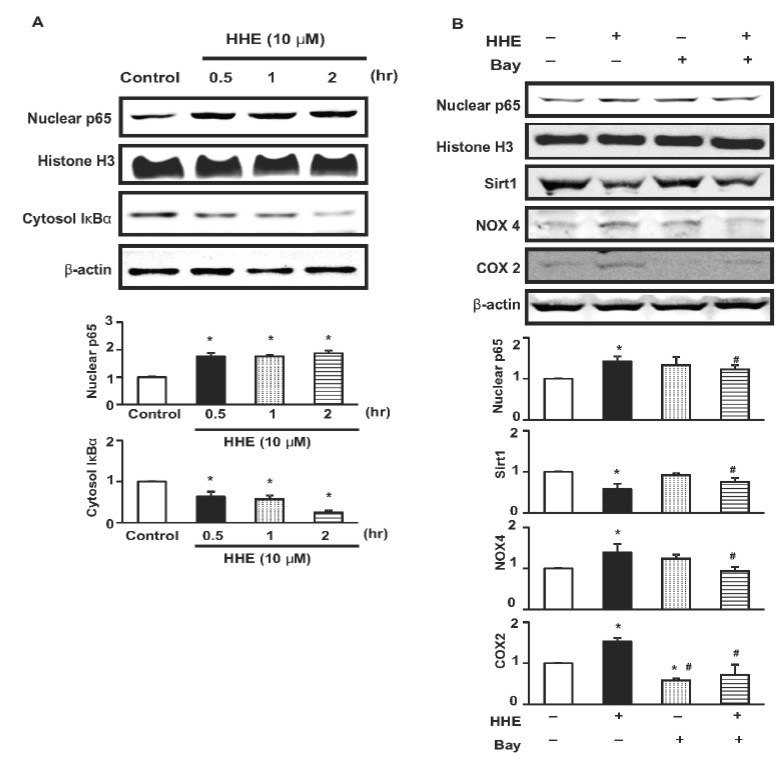 | Fig. 4Expression of NF-κB p65 subunit levels in nuclear extracts of M1 cells incubated with HHE (10 µM) (A). The expression started to increase 30 min after HHE incubation. Cytoplasmic total IκBα expression began to decrease at 30 min, and kept decreased at 1 and 2 hrs. Effects of NF-κB inhibitor (Bay, 10 µM) on expression of NF-κB p65 subunit, Sirt1, NOX4 and COX2 (B). The HHE-induced increased expression of NF-κB p65 subunit in nuclear extracts of M1 cells was also attenuated by 1 hr pre-treated Bay 117082 (10 µM), NF-kB inhibitor. Results are presented as means±SEM of three individual experiments. *p<0.05 vs. control. #p<0.05 vs. HHE.
|
As expected, the observed HHE-induced increase in the nuclear levels of the NF-κB p65 subunit was attenuated by pre-treatment of M1 cells with the NF-κB inhibitor Bay 117082 (10 µM) for 1 hr (Fig. 4B). Notably, however, M1 cells subjected to the HHE/Bay 117082 co-treatment also exhibited reduced expression of Sirt1, NOX4, and COX2 (Fig. 4B).
Analysis of the effects of RSV on M1 cells treated with HHE
Pre-treatment of M1 cells with RSV for 1 hr resulted in abrogation of the observed HHE-mediated increase in nuclear localization of the NF-κB p65 subunit, as well as recovery of the HHE-mediated decrease in cytosolic expression of IκBα (Fig. 5A). Furthermore, the decreases in Sirt1 and AQP2 expression, as well as the increases in NOX4 and COX2 expression, observed upon incubation with HHE (10 µM) were attenuated by RSV treatment (Fig. 5B). HHE treatment resulted in statistically significant increases in the levels of the activated forms of ERK, JNK, and p38 MAPK (pERK, pJNK, and p-p38, respectively). These increases were abrogated by RSV treatment (Fig. 5C). Lastly, HHE treatment resulted in significant increase in the level of Nrf2 and decrease in the level of Keap1, which was counter-regulated by RSV co-treatment (Fig. 5D).
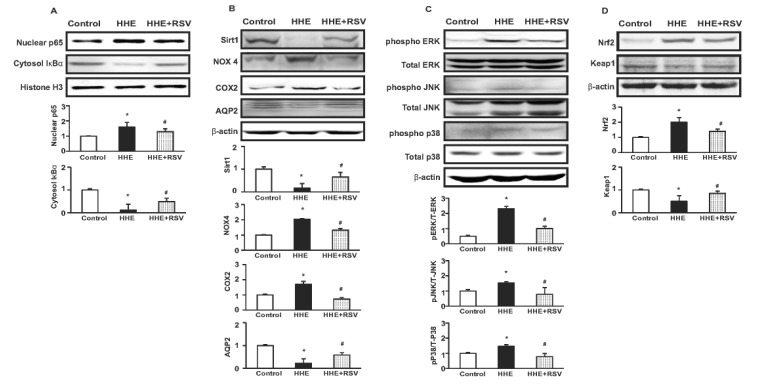 | Fig. 5Effects of resveratrol (RSV) in HHE-treated M1 cells.The protein expression of NF-κB p65 subunit was attenuated and cytoplasmic total IκBα expression was upregulated by resveratrol treatment in M1-cells (A). The protein expression of Sirt1 and aquaporin 2 (AQP2) was decreased by HHE, while NOX4 and COX2 protein expressions were increased. These changes were counteracted by RSV (25 µM) 1 h pre-treatment (B). The protein expression of the phosphorylation of extracellular signal-regulated kinase (pERK 1/2), c-Jun N-terminal kinase (pJNK) and pP38 were increased by HHE treatment, which was attenuated by RSV (25 µM) 1 h pre-treatment (C). The protein expression of nuclear factor erythroid 2-related factor 2 (Nrf2) was increased and decreased in Kelch ECH associating protein 1 (Keap1) with HHE (10 µM) treatment, which was counter-regulated by 1 h RSV pre-treatment (D). N*p<0.05 vs. control, #p<0.05 vs. HHE.
|
Go to :
DISCUSSION
In this study, the observed HHE-mediated decrease in Sirt1 expression was rescued upon co-treatment with Bay 117082, NAC, or RSV. Furthermore, RSV treatment resulted in inhibition of the HHE-mediated increase in NF-κB and MAPK activation. These results therefore suggest that targeting Sirt1 with pharmacologic activators might improve renal function in patients with conditions that can induce oxidative stress in renal collecting duct cells.
ROS metabolism has been proposed to play an important role in all known types of programmed cell death [13]. Specifically, the imbalance between cell survival and death, a key feature of many degenerative and inflammatory diseases, may be caused by aberrant turnover of ROS, a process that in turn regulates the crosstalk between NF-κB and MAPKs [14]. MAPK pathways are a series of parallel cascades of serine/threonine kinases that are activated by diverse extracellular, physical, and chemical stresses and have been shown to regulate cell proliferation, differentiation, and survival [15]. The three major MAPK pathways terminate with the ERK, p38, and JNK/SAPK kinases. The ERK pathway is typically activated by extracellular growth factors, and activation of this pathway has been linked to cell survival. Meanwhile, the p38 and JNK/SAPK pathways are activated by a variety of stresses, including oxidants, UV irradiation, hyperosmolality, and inflammatory cytokines, and have been linked to cell death [16]. In the present study, there was an increase in the levels of activated pERK, pJNK, p38, and ROS generation upon treatment with HHE, suggesting that HHE-induced oxidative stress results in activation of these MAPKs in M1 cells.
The Keap1-Nrf2 pathway is the major regulator of cytoprotective responses to endogenous and exogenous stresses caused by reactive oxygen species (ROS) [17]. The key signaling proteins within the pathway are the transcription factor Nrf2 that binds together with small Maf proteins to the antioxidant response element (ARE) in the regulatory regions of target genes, such as glutathion synthesis [18]. This study showed increased expression level of Nrf2 and decreased expression level of Keap1, which suggest protective mechanism of Keap1-Nrf2 pathway against HHE-induced oxidative stress.
Within the cytoplasm, NF-κB is bound and inactivated by a member of the IκB family of protein inhibitors [19]. Activation of NF-κB is mediated, at least in part, by the phosphorylation (i.e., activation) of ERK, p38, and JNK. Upon activation, these MAPKs promote the phosphorylation and degradation of IκB and the subsequent nuclear translocation of NF-κB [20], resulting in the transcriptional expression and trans-activation of a number of downstream genes [1921]. In the present study, HHE treatment resulted in increased nuclear localization of the p65 subunit of NF-κB and decreased cytoplasmic expression of IκBα. Conversely, RSV treatment attenuated both the nuclear localization of the p65 subunit and the degradation of IκBα. These findings suggest that HHE induces NF-κB activation by stimulating IκBα degradation, and that this process was inhibited by RSV treatment. Furthermore, RSV treatment attenuated the HHE-mediated increase in the levels of pERK, pJNK, and pP38. These findings indicate that the observed RSV-mediated decrease in NF-κB activity may be due to decreased activation of the ERK, JNK, and P38 signaling pathways in M1 cells. In the present study, NF-κB inhibitor treatment restored HHE-induced downregulation of Sirt1 in M1 cells. However, the underlying mechanism remains to be determined. NF-κB, one of the downstream targets for Sirt 1 [2223], is an important transcription factor. Sirt1 could directly interact and deacetylate the p65 component of NF-κB [22] and inhibit NF-κB activity [24]. Taken together, it can be speculated that restored Stri1 expression by NF-κB inhibitor may represent the negative feedback of NF-κB.
Apical plasma membrane localization of AQP2 results in enhanced apical permeability of collecting ducts to water. By controlling the plasma membrane insertion, as well as regulating the transcriptional and post-transcriptional expression of AQP2, arginine vasopressin (AVP) plays a major role in regulating water reabsorption [25262728]. In addition, AQP2 expression is influenced by numerous AVP-independent factors, including hormones, such as aldosterone and insulin [2930], extracellular calcium [31], and environmental tonicity [323334]. Furthermore, it has been revealed that the strong NF-κB activation observed in renal inflammatory diseases most likely accounts for the observed decrease in AQP2 expression and the accompanying urinary concentration defects [35]. In the present study, we demonstrated that AQP2 expression decreased upon treatment with HHE, and that this effect was counter-regulated by RSV treatment. These findings therefore suggest that the RSV-mediated rescuing of AQP2 expression was due, at least in part, to reduced NF-κB/MAPK signaling in M1 cells.
The antioxidant function of Sirt1 has previously been reported in other tissues, including cardiac tissues, neurons, pancreatic β cells, and the interstitial cells of the renal inner medulla [363738]. Given the reported anti-stress and anti-aging functions of Sirt1 [39], treatment with Sirt1 activators may have the potential to protect the kidneys from oxidative stress-related renal diseases. The reported mechanisms by which Sirt1 regulates the cellular response to oxidative stress include modulation of the expression and/or activity of the cell cycle control protein FOXO, the DNA damage repair protein Ku70, and the proteins involved in apoptosis, p53 and E2F1 [40]. Here, we showed that HHE treatment resulted in upregulation of NADPH oxidase and activation of NF-κB and MAPKs, as well as downregulation of Sirt1 in M1 cells, which might contribute renal tubular inflammation. Conversely, the RSV-mediated activation of Sirt1 counteracted this increased expression of NADPH oxidase and oxidative stress markers.
In summary, our findings support an important role for Sirt1, the expression of which is modulated by oxidative metabolism, in protecting M1 cells under oxidative stress conditions. Moreover, the results of this study suggest that treatment of patients with Sirt1 activators to enhance Sirt1 expression may comprise a therapeutic strategy for minimizing or preventing renal damage resulting from increased oxidative stress.
Go to :
 XML Download
XML Download