

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Renal cancer is the most frequent and lethal urological malignancy in adults [1]. Approximately 20~30% of patients with renal cancer present with locally advanced or metastatic disease at the time of diagnosis. Five-year survival of patients with metastatic renal cancer is less than 10% [2]. Although surgical resection can be curative in localized renal cancer, the prognosis of patients is extremely poor and renal cancer is largely resistant to radiation and chemotherapy [3].
Increasing evidences have demonstrated that the development of renal cancer is linked to hyperactivation of tyrosine kinase receptors, Akt/mTOR signaling, and Wnt/β-catenin signaling [45]. Tyrosine kinase inhibitors (TKIs) have provide efficacious therapies for segmental patients with renal cancer, of which some also inhibit p21 [6] and mammalian target of rapamycin inhibitors [7]. However, their effect appears to be rather limited, with only 10~30% of cases exhibiting a complete or partial response. The progression-free survival for these patients is still short, at only 1~2 years [8]. Thus, new pharmacological targets and development of effective drugs for metastatic renal cancer are needed.
Chromosome maintenance protein 1 (CRM1), a ubiquitous nuclear export receptor protein, involves in many intracellular pathways [910]. It can recognize hydrophobic, leucine rich nuclear export signal (NES) of cargo proteins [11] and transport over 200 proteins including transcription factors, tumor suppressor proteins and cell growth regulators such as p53 [12], p21 [10], p27 [13], Foxos [10], and nucleophosmin-1 (NPM1) [14]. By inhibiting CRM1 function, tumor suppressor proteins could be retained in the nucleus, inducing cancer cell apoptosis or cell cycle arrest [10]. More recently, it has been reported that knockdown of CRM1 expression arrests cell cycle progression and inhibits the proliferation of renal cancer cells both in vitro and in vivo [1516]. In addition, multiple studies also showed that CRM1 inhibition suppresses tumor growth in other several cancer cell lines [17181920]. Therefore, the development of drugs targeting CRM1 may provide therapeutic benefit for patients with renal cancer.
A large number of CRM1 inhibitors have been investigated. However, most of them are irreversible inhibitors which have toxicity on normal cells. Leptomycin B (LMB) is the first identified CRM1 inhibitor but is not entered clinical use due to non-tolerated toxicities [21]. Thus, searching for novel compounds, with reduced toxicities that could target nuclear export, is urgently needed. Recently, it has been reported that novel selective inhibitors of nuclear export (SINEs) could significantly inhibit growth of renal cancer cells in vivo [15]. SINE compound (KPT-330) is generally well tolerated and is currently in phase I/II clinical trials for advanced hematologic malignancies and solid tumors [22]. Furthermore, CBS9106, a novel reversible CRM1 inhibitor, was recently identified and reported to have anti-tumor activity against multiple myeloma [23]. We believe that reversible inhibitors of CRM1 may be safer, less toxic, and well-tolerated in patients.
In this study, we reported the effect of a novel reversible CRM1 inhibitor S109 on renal cancer in vitro. S109 is more easily and cost-effectively synthesized derivative of CBS9106. Thus, it should be more suitable for commercial production. We found that S109 can reversibly inhibit the nuclear transport of tumor suppressor proteins mediated by CRM1, resulting in renal cancer cell growth inhibition and cell cycle arrest. Our study suggests that further evaluation of S109 may be hopefully translated to clinical application on renal cancer.
METHODS
Cell culture, antibodies and reagents
Human renal cancer cell line 786-O and OS-RC-2 were purchased from Shanghai Cell Bank of the Chinese Academy of Sciences (CAS), and were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 µg/mL streptomycin. The cells were incubated in a humidified incubator with 5% CO2 at 37℃. Compound S109 was synthesized by Suzhou Komanda Drug Development Company. S109 was dissolved in dimethyl sulfoxide (DMSO) to make a 10 mmol/L stock solution and then was diluted with culture medium at different concentrations before use.
Antibodies to CRM1, RanBP1 and p53 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies to Foxo1, p21, p27, Cyclin D1, and β-actin were purchased from Cell Signaling Technology (CST, Beverly, MA). Alexa 488-conjugated donkey anti-rabbit antibody was purchased from Invitrogen Life Technology (Invitrogen, Carlsbad, CA).
Cell viability assay
Cell viability was evaluated by a Cell Counting Kit-8 (CCK8) assay following the manufacturer's instructions. 786-O cells were harvested and seeded in 96-well plates. After 24 h, the cells were treated with different concentrations of S109 (0.04, 0.2, 1, 5, 25, and 50 µmol/L) and then cultured for 72 h. Control groups received the same amount of DMSO. At the time of collection, 10 µL of CCK8 was added to each well and then incubated for 2 h at 37℃. The optical density (OD) was measured at 450 nm using the microplate reader.
Immunofluorescence
Approximately 5×103 cells were seeded onto 96-well culture plates. Cells were treated with S109 (0.5, 1, 2, and 4 µM) or 0.1% DMSO (vehicle) for 2 h. Then 786-O cells were washed in PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. The fixed cells were permeabilized with 0.3% Triton-X 100 in PBS for 20 min and blocked with 1% bovine serum albumin (BSA) in PBS for 1 h followed by primary antibody (anti-RanBP1) treatment overnight at 4℃. Fluorescent secondary antibody anti-rabbit Alexa 488 was used at 1 : 200 dilution. Nuclei were counterstained with DAPI. The stained cells were visualized on a fluorescence microscopy and photographed (Olympus, Japan).
Cell proliferation and clonogenic assay
The 786-O cells were treated with 0.1% DMSO (vehicle) or S109 at various concentrations (1, 2, and 4 µM) for 12 h. Then, cellular proliferation was evaluated by 5-ethynyl-2'-deoxyuridine (EdU) fluorescence staining using the Cell-Light™ EdU DNA Cell Proliferation Kit (Ruibo Biotech, Guangzhou, China) according to our previous report [24].
To examine the long-term effects, 786-O cells were seeded in six-well plates (600 cells/well) and treated with 0.1% DMSO (vehicle) or S109 (1, 2, and 4 µM) for 12 h. After treatment, the drug-containing medium was removed and fresh medium was added to the wells. Medium was changed every 4 d for 10–14 d to allow for colony formation. Colonies were stained with a crystal violet solution (Sigma Aldrich) and counted manually.
Cell cycle assay
For the cell cycle analysis, 786-O cells were treated with or without S109 for 24 h. Then, the cells were collected, fixed in 70% ethanol, washed twice with PBS and lastly stained with PI solution that contained 25 µg/mL Rnase and 50 µg/mL PI in the dark for 30 min. Subsequently, the cells was assayed on a FACSCalibur (Becton-Dickinson) and analyzed by CellQuest Pro software (Becton-Dickinson).
Establishment of stable expression cell line
The wild type or C528S mutation CRM1 were cloned into a pWPXL lentiviral vector containing a sequence coding for a flag tag. The construct vectors (pWPXL-CRM1 or pWPLX-CRM1-C528S) were co-transfected with packaging vectors into 293T cells using Lipofectamine 2000 (Invitrogen). The supernatant was collected and concentrated after 48 h incubation. 786-O cells were infected with CRM1-WT or CRM1-C528S lentivirals, respectively. After 72 h infection, the cells were continuously cultured in medium containing 2.5 µg/mL puromycin.
Western blot analysis
786-O cells were treated with different concentrations of S109. After 24 h incubation, total protein or nuclear protein extracts from treated or untreated cells were subjected to Western blot analysis as described elsewhere [2526]. The expression patterns of CRM1, Cyclin D1, p53, p21, p27, and Foxo1 were detected using specific antibodies all, β-actin and Histone-H3 were used as the loading control.
RESULTS
S109 inhibits the growth and CRM1-mediated nuclear export in renal cancer cells
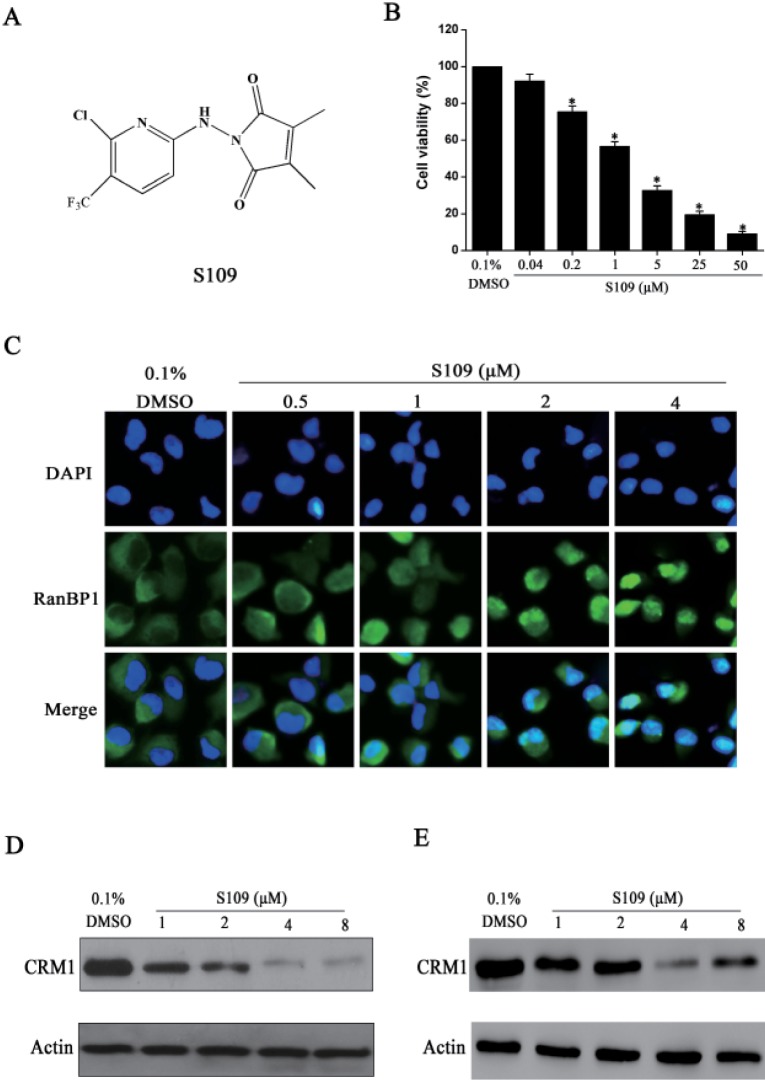
To assess the anti-cancer effect of S109, we examined the viability of 786-O cells using CCK8 assay. As shown in Fig. 1B, these results suggested that cell viability of S109-treated groups were decreased in a dose-dependent manner, compared with control group. The IC50 value of S109 was 1.16 µM for 786-O cells. These results reveal that S109 can significantly inhibit the growth of renal cancer 786-O cells.
Next, we investigated the effect of S109 on CRM1-depentent nuclear export. RanBP1, a Ran-binding protein, shuttles between the nucleus and cytoplasm, but is predominantly localized to the cytoplasm under normal culture conditions. Previous studies have suggested that nuclear accumulation of RanBP1 protein is used as a canonical marker for CRM1 inhibition [27]. As shown in Fig. 1C, RanBP1 is found exclusively in the cytoplasm in vehicle-treated cells. In contrast, treatment with S109 led to nuclear accumulation of RanBP1 in dose-dependent manners. In addition, we also examined the effect of S109 on the expression level of CRM1 protein in 786-O and OS-RC-2 cells. The results showed that the level of CRM1 protein expression was markedly down-regulated on treatment with 4 µM S109 (Fig. 1D and 1E), implying that inhibition of CRM1 activity by S109 may result from the reduction in CRM1 protein expression.
The inhibitory effect of S109 on CRM1 function is reversible
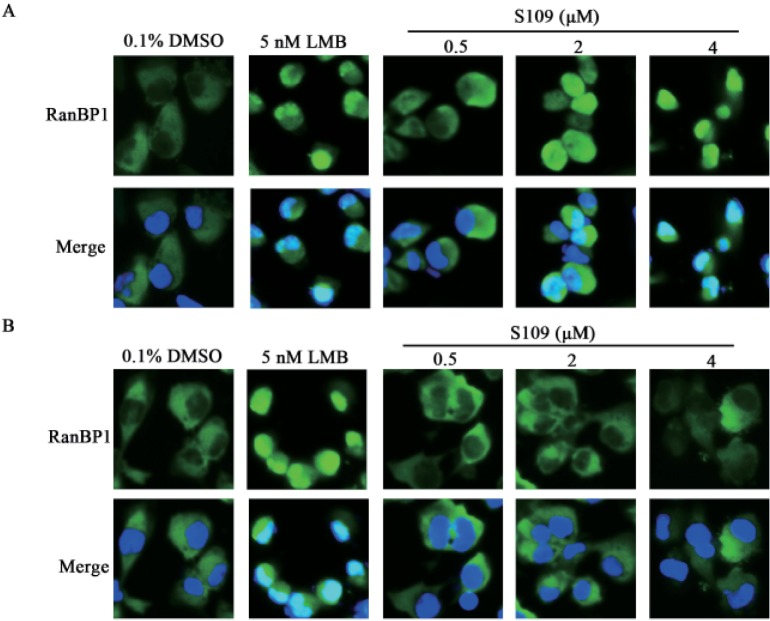
It is well known that LMB irreversibly binds to CRM1 [21]. To investigate whether the inhibition of CRM1 by S109 is reversible, we examined the subcellular localization of RanBP1 after removal of the compound. As seen is Fig. 2A, we incubated 786-O cells with LMB or S109 for 2 h, and then found a strong accumulation of RanBP1 in the nucleus. However, RanBP1 was still in the cytoplasm of untreated cells. The cells were then washed with PBS to remove the LMB or S109, and added new medium for another 2 h incubation. RanBP1 in S109-treated cells was almost completely translocated to the cytoplasm, whereas in LMBtreated cells, RanBP1 was still mainly localized in the nucleus (Fig.2B). Our results preliminarily indicate that the inhibitory effects of S109 on CRM1 function is reversible.
S109 suppresses the proliferation and clone formation of 786-O cells
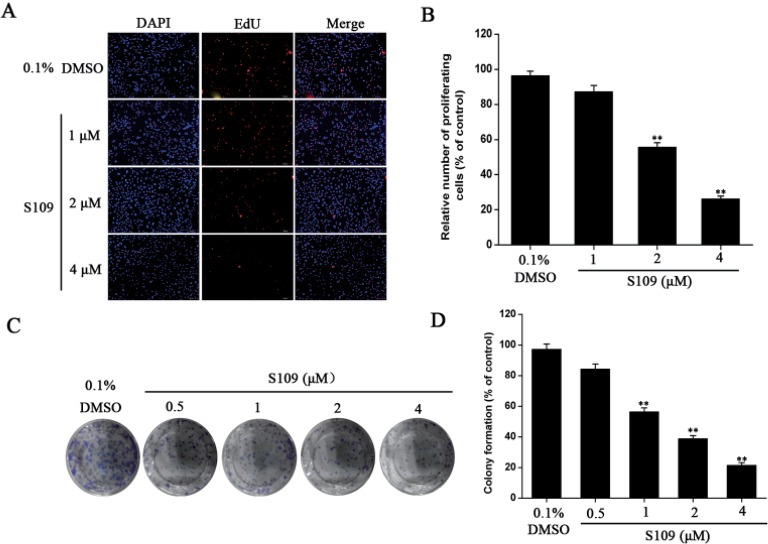
To further assess the effect of S109 on 786-O cell growth, we examined the rates of cell proliferation by EdU fluorescence staining and clonogenic assay. Compared with control group, the number of EdU-positive cells in the S109-treated group was significantly reduced. 786-O cells exposure to 2 and 4 µM S109 reduced the proliferation to approximately 54.28% and 25.37%, respectively (Fig. 3A and 3B). In addition, the clonogenicity of 786-O cells in the S109-treated groups was reduced in a concentration-dependent manner. Compared with the control group, the colony formation markedly decreased by 15.63%, 43.52%, 61.21% and 74.63% in response 0.5, 1, 2, and 4 µM treatment, respectively. These results were consistent with the CCK-8 assay, suggesting that S109 inhibits the proliferation of renal cancer cells.
S109 induces cell cycle arrest via nuclear localization of tumor suppressor proteins
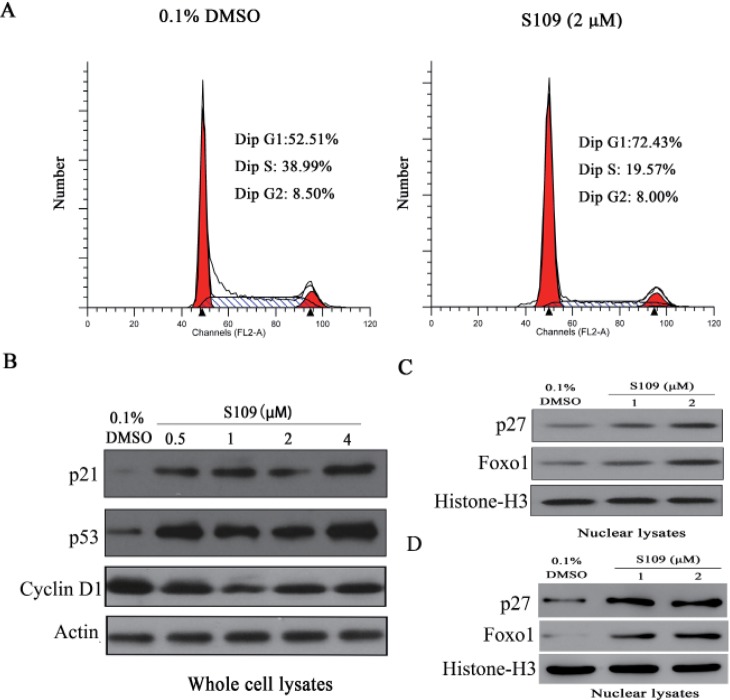
To investigate whether S109 inhibits 786-O cell growth by abrogating cell cycle progression, cell cycle distribution was evaluated using flow cytometric analysis. As shown in Fig. 4A, compared with the control group, the cells treatment with S109 caused a increase of the percentage of cells in the G1 phase, and an clear decrease of the percentage of cells in the S phase. Next, we assessed the effects of S109 on cell cycle related regulating factor. Western blot analysis revealed that S109 inhibited the expression of Cyclin D1 and promoted the expression of p53 and p21 at protein level in a dose-dependent manner (Fig. 4B). Taken together, these results suggest that S109 induces cell cycle arrest in 786-O cells.
Multiple studies have reported that CRM1 inhibitors induce tumor cell growth inhibition by blocking nuclear export of tumor suppressor proteins [13192128]. Therefore, we further examine the effects of S109 on the nuclear accumulation of tumor suppressor proteins in 786-O and OS-RC-2 cells. As shown in Fig. 4C and 4D, S109 treatment resulted in a dose-dependent increase in the nuclear fraction of major tumor suppressor proteins, Foxo1 and p27. These results imply that inhibition of cell cycle progression by S109 may results from the nuclear accumulation of these major tumor suppressor proteins.
Mutation of CRM1 abolishes S109-mediated nuclear export
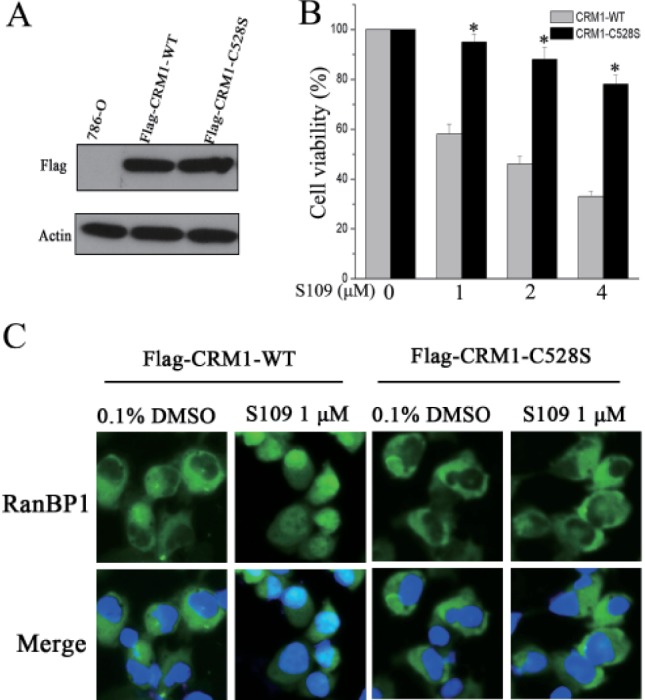
It has been reported that LMB covalently binds to a cysteine residue (Cys528) of CRM1 and inhibits CRM1-mediated protein export. To further prove that S109 regulates protein nuclear export by CRM1 inhibition, we analyzed the subcellular localization of RanBP1 in cells co-expressing either wild-type or C528S mutant CRM1 upon treatment with LMB or S109. As shown in Fig. 5A, the exogenous CRM1 expressed very well immunoblotted with anti-Flag, whereas no expression was monitored in cells of non-transfected lentivirals, indicating that the stable CRM1-WT or CRM1-C528S over-expression 786-O cells was generated successfully.
We then evaluated the effect of CRM1 mutation on cell proliferation in response to S109 treatment. As shown in Fig. 5B, S109 did not significantly inhibit the growth of cells that stably expressed Cys528 mutant CRM1. We further evaluated whether S109 loses its ability to inhibit the nuclear export of RanBP1 in CRM1-C528S over-expressing cells. Consistent with our previous results, S109 can inhibit nuclear export of RanBP1 in CRM1-WT over-expression cells. However, cells expressing Cys528 mutant CRM1 were resistant to S109 treatment, because RanBP1 remained in the cytoplasm (Fig. 5C). The above results suggest that inhibition of CRM1 function by S109 are dependent on binding to the Cys528 of CRM1.
DISCUSSION
Renal cancer is the most fatal among urological malignancy [29]. Current chemotherapeutics can only increase the overall survival of patients from weeks to months and cannot cure renal cancer [29]. Thus, there is an urgent need for newer and more effective drugs for renal cancer treatment, especially for the patients with locally advanced or metastatic disease. In this study, we investigated the antitumor activity of S109 in renal cancer. We found that S109 inhibits the nuclear export of major tumor suppressor proteins mediated by CRM1, ultimately resulting in cell cycle arrest, inhibition of cell proliferation and clonogenic potential of renal cancer cells.
Protein shuttling between the nucleus and the cytoplasm plays an important role in cell proliferation and survival. Some of tumor suppressors must localize in the nucleus to perform their functions. Nuclear export of these key proteins significantly influences treatment outcome [1030]. The over-expression of CRM1 has been found in various tumor, including renal cancer [15], pancreas cancer [18], gliomas [20], and multiple myeloma [23] and is closely associated with poor prognosis. Multiple studies have demonstrated that anti-tumor activity of CRM1 inhibitors in these human solid and hematologic malignancies [171931]. CRM1 over-expression facilitates nuclear export of tumor suppressor proteins, leading to infinite proliferation of tumor cell [1032]. Therefore, CRM1 is deemed to be as a promising therapeutic target for anticancer drug development against renal cancer.
Recently, some small molecule CRM1 inhibitors have been investigated. However, most of them are irreversible inhibitors. In our study, we demonstrated that S109 induces nuclear accumulation of RanBP1, a useful biomarker of CRM1 inhibitor. Most importantly, our results demonstrated that S109 is a reversible CRM1 inhibitor in renal cancer cells. In addition, we also found that S109 reduces CRM1 protein levels, whereas the irreversible inhibitor LMB could not induce decrease of CRM1 [31]. The mechanism of S109 induces decrease of CRM1 is currently unknown. We speculated that the reversible CRM1 inhibitor dissociate from CRM1 may change the conformation of CRM1 protein, and then these changes make the CRM1 more conducive to be recognized by ubiquitin/proteasome system. Mutation of CRM1 at Cys528 position abolishes nuclear export inhibition by S109, these findings indicate that S109 is dependent on binding to the Cys528 of CRM1.
Akt/mTOR signaling pathway plays an important role in oncogenic processes including cell proliferation, survival and angiogenesis [33]. Components of this pathway, specifically Akt, mTOR and p70S6K are constitutively activated in renal cancer compared to normal renal tissue [34]. Foxo1 is an important downstream target effector of Akt/mTOR signaling pathway and is closely associated with cell cycle progression [35]. In this study, we found that S109 induces cell cycle arrest at G1 phase. However, the sub-G1 fraction was not detected. These data suggest that S109 may inhibit cell cycle arrest in renal cancer cells without causing significant apoptosis. It is well known that Cyclin D1 is a nuclear protein required for cell cycle progression in G1, the synthesis of which is initiated during G1 and drives the G1/S phase transition [36]. S109 could down-regulate the expression of Cyclin D1, and induce nuclear accumulation of Foxo1 and p21. These results are strongly correlated with the altered cell cycle distribution phenotype, suggesting that antitumor potential of S109 is due to the restriction of Foxo1 and p27 localization to the nucleus, where they arrest the cell cycle.
In summary, our results show that S109 is a novel CRM1 inhibitor that acts by abolishes CRM1-mediated nuclear export of several tumor suppressor proteins. This leads to proliferation inhibition and cell cycle arrest in renal cancer in vitro. However, further in-depth studies are needed to confirm its anticancer effect in vivo. Based on our present results, we believe that S109 may be as a novel anticancer drug candidate for the treatment of renal cancer.
 XML Download
XML Download