

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Excessive glutamate causes neural dysfunction and degeneration. Excess entry of Ca2+ and the consequent overload of Ca2+ in neuronal cell are an early key step in the excitotoxicity induced by an exposure to excess glutamate in ischemia, epilepsy, and trauma [123]. In addition, Ca2+ signals are propagated from the cytosol into the nucleus [456], regulate expression of some genes [78] and induce neuroprotection [9].
The stimulation of glutamate receptors has been reported to be required for preconditioning in neurons [1011]. It has been reported that NMDA preconditioning is able to prevent quinolinic acid-induced neurotoxicity and seizure [1213]. Calcium signals induced by activation of the glutamatergic receptor during preconditioning may protect against neuronal cell death [14]. Preconditioning by Ca2+ ionophore ionomycin has been reported to decrease oxygen-glucose deprivation (OGD)-induced cell death [15].
A reduction in extracellular Mg2+ concentration ([Mg2+]o) to 0.1 mM in neurons elicits an intense pattern of [Ca2+]i spikes [161718192021]. The low [Mg2+]o-induced Ca2+ spikes are synchronized in an active network and induce neuronal cell death [182122]. In addition, the low [Mg2+]o in neuronal cells or slice has been characterized as an in vitro seizure model [202324]. However, little is known about whether the synaptically-induced Ca2+ spikes by low [Mg2+]o directly induce preconditioning in cultured hippocampal neuronal cells.
In the present study, we examined whether hippocampal neurons that are preconditioned by a brief exposure to low [Mg2+]o are rendered resistant to subsequent prolonged exposure-induced neuronal cell death 24 h later and whether Ca2+ signals are involved in this process. Low [Mg2+]o preconditioning induced excitotoxic tolerance through the [Ca2+]i spike-induced activation of PKCε and PKCζ, JAK-2, MAPK kinase, CaMKII and the de novo synthesis of proteins.
METHODS
Materials
Materials were purchased from the following companies: fura-2 acetoxymethyl ester (AM) from Molecular Probes (Eugene, OR, USA); Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) from Invitrogen (Carlsbad, CA, USA); the AMPA receptor antagonist 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo [f] quinoxaline-7-sulfonamide (NBQX), the L-type Ca2+ channel antagonist nimodipine, PKCε translocation inhibitor and PKCζ pseudosubstrate inhibitor, CaMKII inhibitor KN-62 from Calbiochem (San Diego, CA, USA); the NMDA receptor antagonist D-2-amino-5-phosphonovalerate (D-AP5), the intracellular calcium chelator BAPTA, 1,2-bis(2-aminophenoxy)ethane-*N,N,N',N'-tetraacetic acid; the nonspecific PKC inhibitor staurosporin, the JAK-2 inhibitor AG490, the MAPK kinase inhibitor PD98059 and all other reagents from Sigma (St. Louis, MO, USA). Mg2+ free-DMEM (DMEM without MgSO4) was purchased from WelGENE (Korea) as a customer-ordered item.
Cell culture
Embryonic day 17 hippocampal neurons from adult maternal Sprague-Dawley rats (250~300 g) were grown in primary culture as described previously [21]. All procedures performed on the animals were conducted with the approval of the Catholic Ethics Committee of the Catholic University of Korea. Cells were pelleted and resuspended in 0.814 mM [Mg2+]o Dulbecco's modified Eagle's media (DMEM) without glutamine and supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (100 U/ml and 100 µg/ml, respectively). Dissociated cells were then plated at a density of 50,000 cells/well onto plain 25-mm-round cover glasses (Fisher Scientific) or microetched coverslips (Bellco Biotechnology) that had been coated with poly-L-lysine (0.1 mg/ml). The cells were used between 13.5 and 15.5 days in culture (DIC). During this period, neurons developed extensive neuritic networks and formed functional synapses.
Induction of preconditioning by 0.1 mM [Mg2+]o medium in primary hippocampal neurons
In the 0.1 mM [Mg2+]o medium-induced preconditioning experiments, at least 100 neurons were counted on microetched coverslips [21]. Hippocampal neurons were exposed to 0.1 mM [Mg2+]o medium for 3~5 min and then 24 h later exposed to the same medium for 24 h. 0.1 mM [Mg2+]o DMEM were made by adding 0.1 mM MgCl2 and 0.01 mM glycine into the Mg2+ free-DMEM (WelGENE) just before experiments. Drugs, when included, were added to the 0.1 mM [Mg2+]o medium application simultaneously with the cells and given a 5 min exposure. Viable neurons were identified based on morphological criteria; they were phase-bright, had rounded somata, and extended fine processes [21]. Cell death was determined by comparing the number of viable neurons before and after treatment at the same microscopic fields based on the location of the microetched grid coverslips. Viable neurons from at least 5 randomly selected microscopic field on the location of the microetched grid coverslips were counted and averaged. In some experiments, viability was confirmed by demonstrating that cells identified as viable also excluded propidium iodide (50 µg/ml).
Digital Calcium Imaging
To measure intracellular Ca2+ concentration ([Ca2+]i), hippocampal cells were incubated in 5 µM fura-2 acetoxymethyl ester (AM) (Molecular Probes) in HEPES-buffered Hanks' Salt Solution (HHSS) containing 0.5% BSA for 45 min at 37℃. The Hank's salt solution was composed of (in millimoles per liter): HEPES, 20; NaCl, 137; CaCl2, 1.3; MgSO4, 0.4; MgCl2, 0.5; KH2PO4, 0.4; Na2H2PO4, 0.6; NaHCO3, 3.0; and glucose, 5.6. The cover glass was then mounted in a flow-through chamber [18] that was superfused at a rate of 1.5 ml/min. Digital calcium imaging was performed as described previously [18]. [Ca2+]i spikes were induced by HHSS containing 0.1 mM MgCl2 and 0.01 mM glycine.
RESULTS
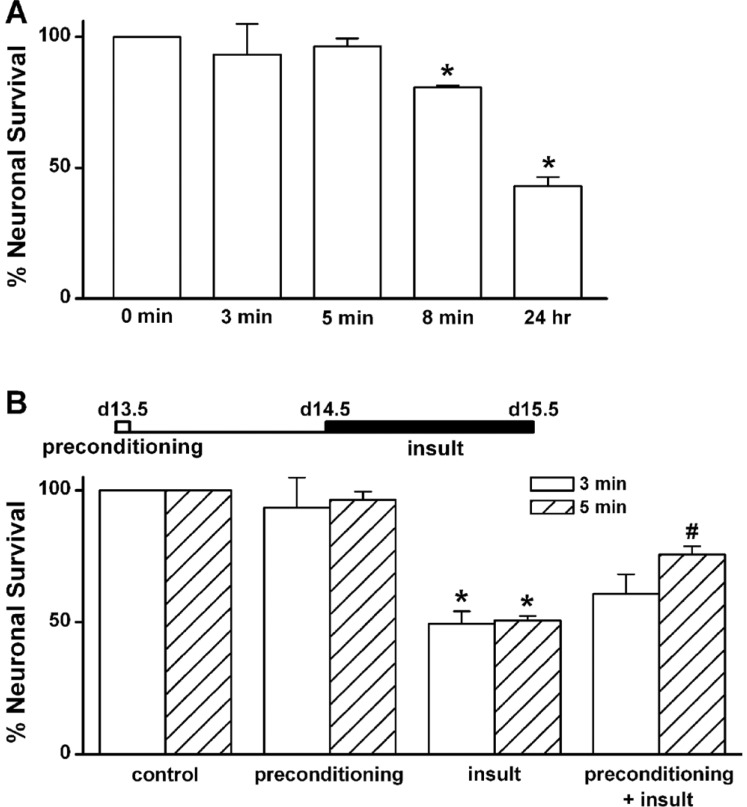
In vitro preconditioning by a brief exposure to low [Mg2+]o inhibits prolonged exposure-induced neuronal cell death in cultured rat hippocampal neurons
We adapted the low [Mg2+]o-induced Ca2+ spiking activity, which produces neuronal cell death when sustained [21], to model in vitro preconditioning. To determine the time of 0.1 mM [Mg2+]o exposure required to induce preconditioning in cultured rat hippocampal neurons, we replaced the normal DMEM containing 0.814 mM [Mg2+]o with 0.1 mM [Mg2+]o DMEM for various time periods at day 13.5 (Fig. 1A). When an exposure to 0.1 mM [Mg2+]o induces [Ca2+]i spikes [19], a brief exposure to the 0.1 mM [Mg2+]o media for 3 or 5 min did not elicit cell death when assayed 24 h later (3 min: 93.4±11.5% of control, n=4; 5 min: 96.4±3.1% of control, n=20, respectively). However, an exposure to the low [Mg2+]o for 8 min slightly elicited cell death (8 min: 79.2±1.7% of control, n=4). Exposure to the low [Mg2+]o for 24 h induced marked neuronal death (43.1±3.4% of control, n=4).
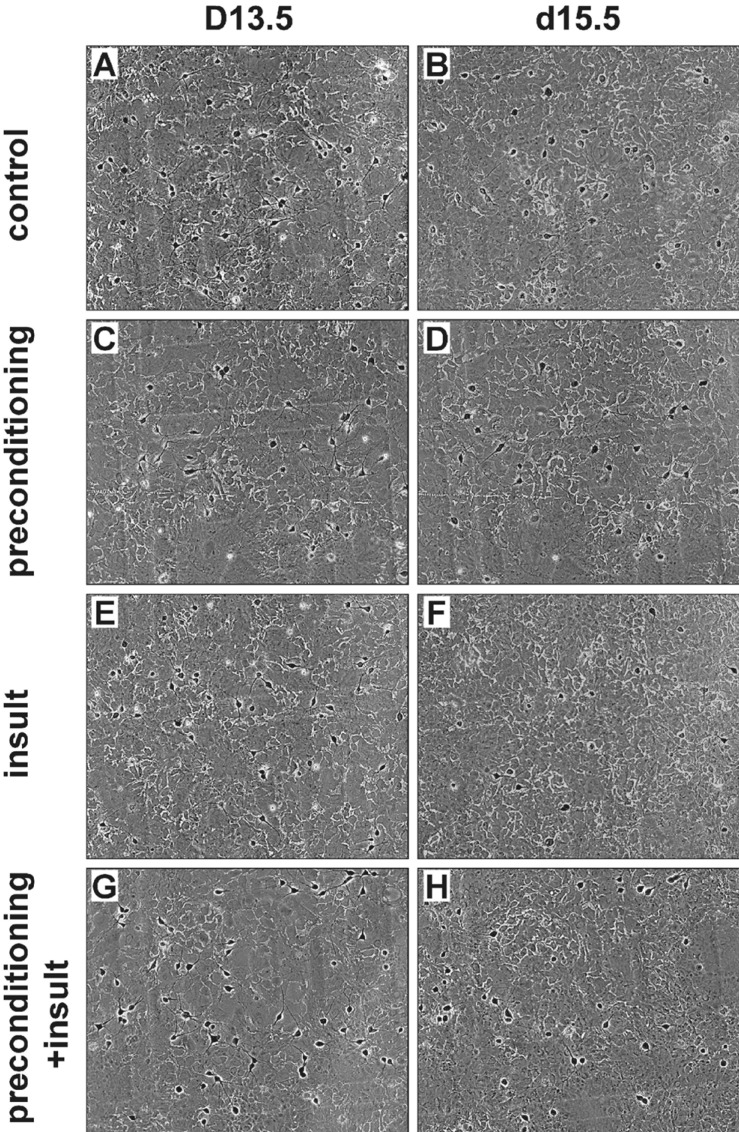
In order to determine whether a brief exposure to 0.1 mM [Mg2+]o protects cells from excitotoxicity induced by a second prolonged exposure to the same insult, cells were preconditioned with 0.1 mM [Mg2+]o medium for 3 or 5 min and then 24 h later exposed to the same medium for 24 h (Fig. 1B). Low [Mg2+]o preconditioning for 3 min did not significantly inhibit the prolonged exposure-induced neuronal death (60.8±7.3%, n=4). However, preconditioning by the exposure for 5 min significantly inhibited neuronal death (75.7±3.0%, n=20). Therefore, we used a paradigm of a 5 min preconditioning and a 24 h main insult to study the characteristics of low [Mg2+]o-induced tolerance in excitotoxicity. Fig. 2 shows representative photographs of effects of the preconditioning on low [Mg2+]o-induced neuronal death. In control cells (medium exchange only), 50.5±1.3% of the cells died (Fig. 2A and B). This value is similar to the previous observations that 50.7±1.7% of control cells may die by extrapolation for 48 h since 22±2% of hippocampal neurons die in a 24 h culture under similar culture conditions [18]. A similar proportion of the preconditioned cells without an insult to control cells disappeared as the result of the preconditioning (Fig. 2B & D). However, the low [Mg2+]o-induced neuronal cell death was markedly inhibited by the preconditioning (insult: 49.4±4.8% of control, n=4; preconditioning + insult: 75.7±3.0% of control, n=4) (Fig. 2F & H).
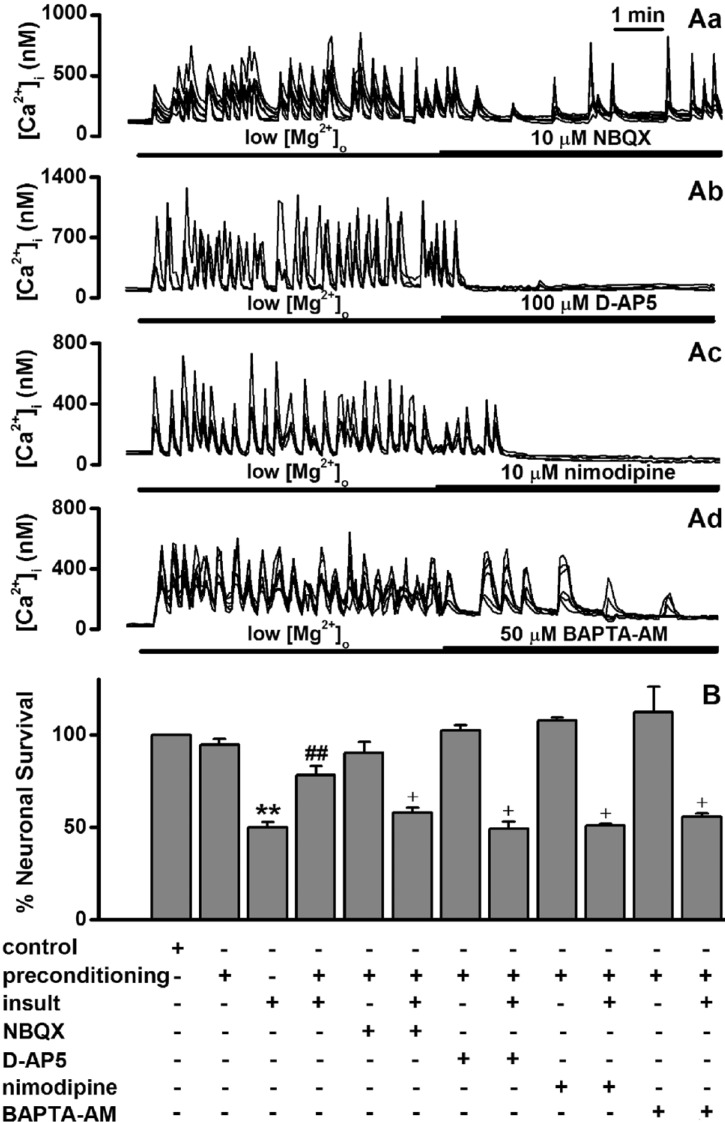
Roles of [Ca2+]i spikes in low [Mg2+]o-induced preconditioning effects
Low [Mg2+]o can elicit an intense pattern of [Ca2+]i spikes that are dependent on glutamatergic synaptic transmission [19]. We examined whether the low [Mg2+]o-induced [Ca2+]i spikes are involved in the low [Mg2+]o preconditioning-induced excitotoxic tolerance. An exposure of hippocampal neurons to 0.1 mM [Mg2+]o induced synchronized [Ca2+]i spikes within 30 s at day 13.5 (Fig. 3A). Treatment with the AMPA receptor antagonist NBQX (10 µM) partly decreased the frequency and amplitude of the [Ca2+]i spikes (Fig. 3Aa). The NMDA receptor antagonist D-AP5 (100 µM) and the L-type voltage-gated Ca2+ channel antagonist nimodipine (10 µM) completely blocked the [Ca2+]i spikes (Fig. 3Ab and Ac). In addition, BAPTA-AM (50 µM) gradually reduced both the frequency and amplitude of the [Ca2+]i spikes (Fig. 3Ad). BAPTA-AM (50 µM) has been reported to slightly inhibit the [Ca2+]i response to higher concentration of glutamate (500 µM) [25].
In subsequent experiments, we determined the effects of these [Ca2+]i changes on low [Mg2+]o preconditioning-induced excitotoxic tolerance (Fig. 3B). Treatment with NBQX during the 5 min period of preconditioning significantly inhibited the tolerance. D-AP5 and nimodipine markedly inhibited the tolerance to a greater extent, respectively. Moreover, BAPTA-AM significantly inhibited the tolerance. All these data are consistent with an evidence that [Ca2+]i spikes are involved in low [Mg2+]o preconditioning -induced excitotoxic tolerance.
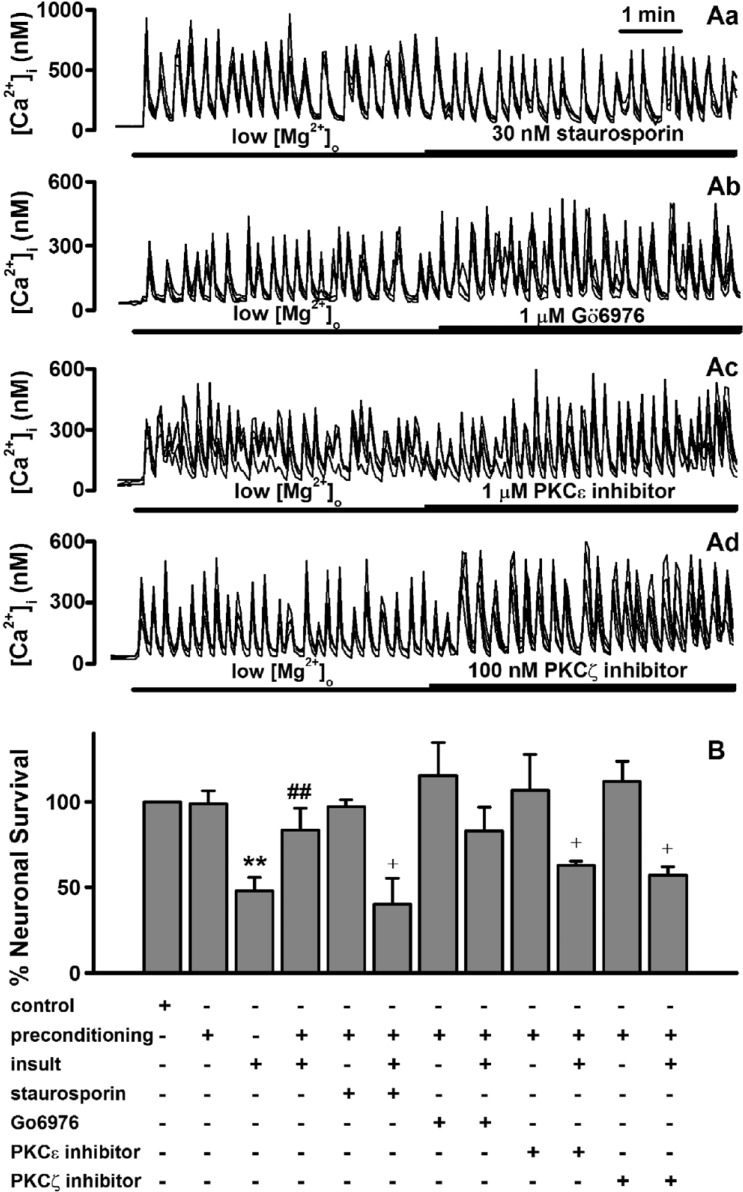
Roles of protein kinase C in low [Mg2+]o preconditioning -induced excitotoxic tolerance
In subsequent experiments, we attempted to determine how low [Mg2+]o-induced [Ca2+]i spikes induce the low [Mg2+]o preconditioning-induced excitotoxic tolerance in hippocampal neurons. An increase in [Ca2+]i can directly and indirectly activate PKC isozymes. PKC has been reported to play a role in brain ischemic preconditioning [142627]. In order to study the role of PKC in low [Mg2+]o preconditioning-induced excitotoxic tolerance, we screened four PKC inhibitors, staurosporin, Gö6976, a PKCε translocation inhibitor, and a PKCζ pseudosubstrate inhibitor, which alone does not affect [Ca2+]i spikes (Fig. 4A). Treatment with staurosporin (30 nM), a non-specific inhibitor of PKC, during the preconditioning significantly reduced the tolerance (Fig. 4B). To investigate which specific PKC isozymes are involved in the preconditioning, the specific PKC inhibitors were used (Fig. 4B). Gö6976 (1 µM), a specific inhibitor of PKCα had no effect on the tolerance. However, both the specific PKCε translocation inhibitor peptide (1 µM) and PKCζ pseudosubstrate inhibitor (100 nM) significantly inhibited the tolerance.
Low [Mg2+]o preconditioning-induced excitotoxic tolerance through activation of other protein kinases
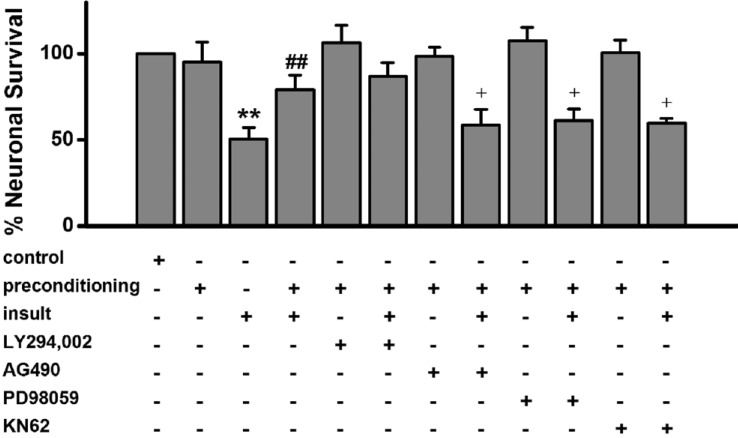
The Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway has been reported to play an essential role in the development of preconditioning in myocardial ischemia [282930], suggesting that the JAK-STAT pathway could be involved in seizure preconditioning in hippocampal neurons. As shown in Fig. 5, treatment with the specific JAK-2 inhibitor AG490 during the preconditioning significantly inhibited the tolerance. However, AG490 alone had no effect on the [Ca2+]i spikes (Data are not shown) as well as on neuronal viability (Fig. 5). Phosphoinositide-3kinase (PI3-kinase) [31], mitogen activated protein kinase (MAPK) kinase [26] and Ca2+/calmodulin dependent protein kinase II (CaMKII) [32] have been reported to reduce ischemic preconditioning effects in an oxygen-glucose deprivation (OGD) model. Treatment with the PI3-kinase inhibitor LY294,002 did not significantly inhibit the tolerance. However, both the MAPK kinase inhibitor PD98059 and the CaMKII inhibitor KN-62 significantly inhibited the tolerance (Fig. 5). LY294002 (5 µM), AG490 (3 µM), PD98059 (3 µM) or KN-62 (5 µM) alone had no effect on [Ca2+]i spikes (Data are not shown) and low [Mg2+]o-induced neuronal death (Fig. 5).
Effects of protein synthesis inhibitor on low [Mg2+]o preconditioning-induced excitotoxic tolerance
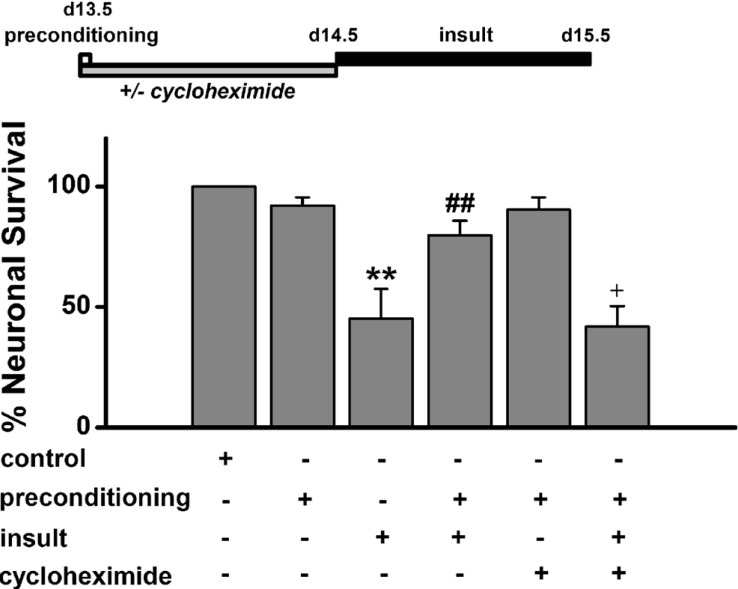
To examine whether the synthesis of de novo proteins is necessary for low [Mg2+]o preconditioning -induced excitotoxic tolerance, we treated cells for 24 h with the protein synthesis inhibitor cycloheximide (0.5 µg/ml) from beginning of preconditioning to immediately prior to prolonged insult in order to block preconditioning-induced protein synthesis (Fig. 6). Cycloheximide abolished the tolerance (41.9±8.5%, n= 6) while it alone had no effect on neuronal survival. This finding indicates that de novo protein synthesis is necessary in low [Mg2+]o preconditioning -induced excitotoxic tolerance.
DISCUSSION
The findings herein show that in vitro preconditioning by a brief exposure to 0.1 mM [Mg2+]o protected cells from subsequent prolonged exposure-induced excitotoxicity in hippocampal neurons. The low [Mg2+]o preconditioning-induced excitotoxic tolerance was suppressed by the inhibition of [Ca2+]i spikes. Furthermore, the tolerance was inhibited by the inhibition of PKCε, PKCζ, JAK-2, MAPK, and CaMKII without affecting the [Ca2+]i spikes during the preconditioning. In addition, the low [Mg2+]o-induced tolerance was blocked by inhibition of de novo protein synthesis.
Although a sustained exposure to low [Mg2+]o for 24 hr is known to be harmful, the present study indicates that a brief exposure to low [Mg2+]o can also lead to adaptations that protect neurons from prolonged exposure to low [Mg2+]o (tolerance). We defined the duration of a brief, nonlethal preconditioning exposure. A paradigm of a 5 min preconditioning and a 24 h main insult was optimal to study the characteristics of the tolerance in our in vitro model. A brief exposure of hippocampal neurons to low [Mg2+]o for 5 min, which induces synaptically synchronized [Ca2+]i spikes, inhibited subsequent prolonged exposure-induced excitotoxicity. The low [Mg2+]o-induced Ca2+ spikes are synchronized in an active network and induce neuronal cell death [182122]. The low [Mg2+]o in neuronal cells or slice has been characterized as an in vitro seizure model [202324]. All these data suggest a possibility that the brief synaptically-induced Ca2+ spikes by seizure-inducing low [Mg2+]o directly induce preconditioning and prevent seizure as well as excitotoxicity. In fact, it has been reported that one or more brief seizure episodes to prolonged seizures render neurons resistant to subsequent more severe insults in vivo [3334] and in vitro [35].
D-AP5 or nimodipine completely inhibited the low [Mg2+]o-induced [Ca2+]i spikes and NBQX significantly inhibited it, thus confirming previously reported results [18]. D-AP5 and nimodipine markedly, and NBQX slightly inhibited the tolerance, which depends on their inhibition of [Ca2+]i spikes. Moreover, the chelation of intracellular Ca2+ with BAPTA-AM inhibited the tolerance. These observations are indirectly supported by other studies that activation of the NMDA receptor is involved in the preconditioning effects [11121426]. This study is directly supported by others' results that the NMDA receptor and Ca2+ are implicated in the induction of anoxia-induced tolerance in cortical slices [36] and ischemia-induced tolerance in hippocampal slices [1415]. Ca2+ spikes have also been reported to induce effectively the expression of specific genes [7]. Moreover, nuclear Ca2+ and the cAMP response element-binding protein family have been reported to mediate a late phase of activity-dependent neuroprotection [9]. Ischemic preconditioning in heart has been reported to be attenuated by the inhibition of voltage-dependent Ca2+ [37]. However, ischemic preconditioning of the cortical neurons was not inhibited by the removal of extracellular Ca2+ or the inhibition of the AMPA receptor [38]. All these data suggest that preconditioning effects are dependent on the preconditioning methods and the location of the tissue used in the study.
Cellular signaling through Ca2+, which is partly mediated by protein kinases such as PKC, CaMKII, and other kinases, has been previously described in ischemic cell death [39]. The intracellular signaling triggered by preconditioning stimuli is achieved by post-translational modifications, primarily the phosphorylation of key proteins [101132]. In this study, inhibition of a novel PKCε or an atypical PKCζ during the preconditioning decreased the low [Mg2+]o-induced tolerance. The translocation of PKC and the phosphorylation of several membrane proteins are mediated through NMDA receptor-mediated Ca2+ influx [40]. In vitro hippocampal neurons express various PKC isoforms including PKCε and PKCζ [41]. Ischemic or NMDA-induced neuroprotection was the results of the activation of PKCε during preconditioning in hippocampal slices [1426] and neuronal cultures [27]. These results suggest that the low [Mg2+]o preconditioning-induced excitotoxic tolerance is mediated through the Ca2+-induced activation of PKCε and PKCζ, although PKC activity has been reported not to be involved in ischemic preconditioning in cortical cultures [38]. Since PKCε and PKCζ are Ca2+-insenstive PKCs, low [Mg2+]o-induced [Ca2+]i spikes may indirectly induce the activation of PKCε and PKCζ during the low [Mg2+]o preconditioning.
Although CaMKII plays a key role in mediating some of the biochemical events that lead to cell death following an ischemic insult [42], treatment with KN-62 inhibited the low [Mg2+]o-induced tolerance without affecting [Ca2+]i spikes in the present study. This observation is directly supported by a report that treatment with KN-62 during ischemic preconditioning suppressed the development of subsequent tolerance in rat cortical cultures [32]. In addition, CaMKII-α knockout mice are more susceptible to cerebral ischemia [43]. These data suggest that the activation of CaMKII is involved in Ca2+-mediated tolerance as well as neuronal cell death. On the other hand, treatment with KN62 during NMDA preconditioning did not significantly reduce the quinolinic acid-induced seizure [13].
JAK signaling is important in ischemic preconditioning in the heart [28]. The inhibition of JAK-2 with AG490 abolishes erythropoietin-induced tolerance against cerebral ischemia [31]. In this study, AG490 inhibited the low [Mg2+]o-induced tolerance. Although the issue of how [Ca2+]i spikes activate JAK-2 remains unknown, these data strongly suggest that JAK-2 is involved in low [Mg2+]o-induced tolerance.
PI3-kinase has been implicated in anti-apoptotic signaling in various cell types, including neurons [44]. LY294,002 did not block the low [Mg2+]o-induced tolerance, consistent with a previous report indicating that PI3- kinase activity is not required for preconditioning in an OGD model [11]. Ischemic preconditioning has also been linked to the phosphorylation of extracellular signal-regulated protein kinases (ERKs) [45]. Ras, Raf, MEK and ERK are required for the development of neuronal preconditioning induced by OGD [11]. Preconditioning by Ca2+ ionophore ionomycin has been reported to increase the MAP kinase p42/44 after OGD [15]. In our study, PD98059 inhibited the low [Mg2+]o-induced tolerance. These observation are supported by in vivo observations of phosphorylation of MEK and ERK in the rat hippocampus in global cerebral ischemic preconditioning [45] and the inhibition of in vitro Mg2+-free preconditioning with U0126 [10]. In contrast to our result, Tauskela et al. [32] reported that treatment with MAPK kinase inhibitors had no effect on OGD-induced preconditioning effects in rat cortical neurons.
Calcium ions have the ability to propagate to the nucleus. The activity of the transcription factors Elk-1, CREB, and MEF-2 is induced by calcium [8]. Moreover, Ca2+ spikes efficiently induce the expression of certain specific genes [7]. These results support the possibility that low [Mg2+]o-induced [Ca2+]i spikes induce the de novo synthesis of proteins. In fact, low [Mg2+]o-induced tolerance was blocked by treatment with cycloheximide, suggesting that the tolerance is dependent on the synthesis of de novo proteins. However the issue of which proteins are synthesized by [Ca2+]i spikes remains to be elucidated. In conclusion, a brief low [Mg2+]o preconditioning induces subsequent prolonged exposure-induced excitotoxicity. The low [Mg2+]o preconditioning-induced excitotoxic tolerance was directly or indirectly mediated through the [Ca2+]i spike-induced activation of PKCε, PKCζ, JAK-2, MAPK kinase, CaMKII and the de novo synthesis of proteins, which is involved in seizure as well as excitotoxicity.
 XML Download
XML Download