

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In general, nitric oxide (NO) plays an important role as a signaling molecule with diverse effects on physiological processes such as cell growth, inflammation, neurotransmission, and bone cell function [1]. NO is a labile free radical that is synthesized via oxidation of L-arginine in a reaction catalyzed by the nitric oxide synthase (NOS) pathway. Three isoforms of NOS are neuronal and endothelial NOS (nNOS and eNOS) are known as constitutive forms. Inducible NOS (iNOS) is the third isoform and is induced by stimulation with microbial products, such as lipopolysaccharide (LPS) and double stranded RNA or pro-inflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) [23]. Cytokine-induced NO causes several activities such as persistent vasorelaxation in septic shock, tumor cell killing, inhibition of cell proliferation, or induction of apoptosis [4].
Osteoblasts play an important role in preservation of bone structure. Bone homeostasis is maintained by bone remodeling, a process that is balanced by osteoblast-mediated bone formation and osteoclast-mediated bone resorption [5]. Dysregulation of the bone remodeling process by systemic and local mediators leads to pathological diseases such as osteoporosis and osteoarthritis [6]. Accumulating evidence suggests that NOS isoforms exist in the bone cells. Expression of iNOS can be induced in osteoblasts in vitro by exposure to pro-inflammatory cytokines and endotoxin [789]. Previous reports demonstrated that MC3T3-E1 cells, osteoblast-like cells, can be induced by pro-inflammatory cytokines and bacterial endotoxin to produce NO [101112131415]. In osteoclasts, lower NO levels appear essential for osteoclastic activity leading to bone resorption, whereas higher NO levels inhibit bone resorption [1617]. It has shown that bone turnover was suppressed by high levels of NO in severe inflammation [9].
Autophagy is known as a self-degradative process that delivers cytoplasmic components to the lysosome [18]. Autophagy plays a role in maintaining cellular homeostasis with degradation of long-lived proteins and damaged intracellular organelles, such as mitochondria, endoplasmic reticulum, and peroxisomes. It is also upregulated to promote cell survival in several stress conditions such as nutrient starvation, pathogen infection, hypoxic condition, and chemotherapeutic agents [19202122]. However, successive autophagy activation can induce cell death by constitutive degradation of important cellular components [23]. The autophagic process includes the formation of double layers of the isolated membrane, sequestering the cargo, and later degrading with fusion of the lysosome to create autolysosomes, resulting in the digestion and ultimate recycling of the compartment [24]. Genetic studies in yeast identified different autophagy-related proteins (ATG), which have specific functions from the initiation to maturation of the process. Among these, LC3 (microtubule-associated protein 1 light chain 3), the mammalian homologue of yeast Atg 8, is involved in the elongation of the phagophore and the formation of the autophagosome. Beclin-1, the mammalian homologue of yeast Atg 6, is also a positive regulator of autophagic vacuole formation [25]. In addition, p62, the ubiquitin-binding scaffold protein that aggregates with ubiquitinated protein, is used as a marker to study autophagic flux. Atg7 protein in the yeast shows homology to the ATP-binding and catalytic sites of the E1 ubiquitin activating enzymes, and is important for the recruitment of proteins to the autophagosomal membrane and the formation of autophagic vacuoles [26].
It is known that autophagy is involved in programmed cell death (PCD). There are many studies assessing the interplay between autophagy and apoptosis in various cells at different levels, including functional and mechanical interaction [27282930]. One aspect of this complexity probably reveals the dual role of autophagy, which is both cell protective and cell destructive depending on different conditions. Several recent studies provided evidence that the stimulation of autophagy during apoptosis can be either a defensive mechanism or a process that contributes to cell death [233031]. The role of NO-induced autophagy in MC3T3-E1 cells has not yet been reported, although recent reports showed that NO in various cells regulates the cross talk between autophagy and apoptosis [32333435]. The objective of this study is to determine the role of NO-induced autophagy in MC3T3-E1 cells and the possible mechanism.
METHODS
Chemicals and reagents
Cell culture media alpha modified Eagle's medium (α-MEM) and fetal bovine serum (FBS) were purchased from GIBCO (Gibco-BRL, USA). Phosphate buffered saline (PBS), sodium nitroprusside (SNP), 3-methyladenine (3MA), compound C, and acridine orange were purchased from Sigma (MO, USA). The primary antibodies used were monoclonal mouse anti-β-actin antibody (Santa Cruz, CA, USA), monoclonal rabbit anti-LC3 antibody, polyclonal rabbit anti-p62 antibody, polyclonal rabbit anti-ATG7 antibody, polyclonal rabbit anti-Beclin-1 antibody, polyclonal rabbit anti-cleaved caspase-3 (Asp175) antibody, polyclonal rabbit anti-PARP antibody, polyclonal rabbit anti-AMPK antibody, and polyclonal rabbit anti-p-AMPK antibody (Cell Signaling, USA).
Cell culture and treatment with SNP
Osteoblastic MC3T3-E1 cells were cultured in α-Minimum Essential Medium (MEM) containing 10% FBS and 1% penicillin-streptomycin reagent (Gibco-BRL). Cell cultures were maintained at 37℃ in a humidified atmosphere of 5% CO2 and 95% air. SNP was dissolved in distilled water and sterilized through a 0.2 µm filter. Cells were treated with each concentration of SNP for the required time period in the same medium.
Cell viability by MTT assay
The MTT assay relies on the observation that viable cells with active mitochondria reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, USA) into a visible dark-blue formazan reaction product and provide an indirect measurement of cell viability. MC3T3-E1 cells were plated onto 24 well plates (5×104 cells/well) and exposed to SNP alone or pretreated with 3MA and SNP for 1 hr. After treatment, MTT was added to the culture medium at a final concentration of 0.1 mg/ml and incubated at 37℃ for 3 hrs. The MTT reaction product was extracted with dimethyl sulfoxide (DMSO, Sigma, USA), and optical density (OD) was measured spectrophotometrically at 570 nm with DMSO as a blank using an absorbance microplate reader (ELx800uv, BioTek, USA).
Trypan blue staining
Cells were plated in 6-well plates and treated with SNP in the absence or presence of compound C. After 24 hrs culture, cells were harvested by trypsinization and stained using a 0.4% trypan blue solution (Gibco-BRL). Trypan Blue positive and negative cells were counted using a hemocytometer and the number of dead cells was calculated as percentage of dead cells on the total number of cells.
Detection of autophagic vacuoles by acridine orange
The acidic autophagic vesicles were visualized by acridine orange staining. Briefly, the MC3T3-E1 (5×104) cells were cultivated on glass chamber slides in α-MEM containing 10% FBS and SNP in the absence or presence of 3MA. At the end of the incubation period, cells were treated with 1 µg/ml acridine orange (Sigma) in serum-free medium for 15 min at 37℃. Subsequently, the cells were washed three times in PBS (acridine orange was removed) and examined under a Confocal Laser Scanning Microscope (Carl Zeiss, Germany). Depending on their acidity, autophagic lysosomes appeared as yellow-orange to bright-red fluorescent cytoplasmic vesicles, while nuclei were stained green.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) staining
TUNEL staining kit (DeadEnd Fluorometric TUNEL system, Promega, USA) was used for detecting DNA fragmentation that results from apoptotic programmed cell death. The assay relies on the presence of nicks in the DNA which can be identified by terminal deoxynucleotidyl transferase or TdT, an enzyme that catalyzes the addition of dUTPs that are secondarily labeled with a marker. It may also label cells that have suffered severe DNA damage. Briefly, the MC3T3-E1 (5×104) cells were cultivated on glass chamber slides in α-MEM containing 10% FBS, and were treated with SNP in the absence or presence of compound C.
Western blot analysis
The MC3T3-E1 cells were washed twice with PBS and proteins were solubilized in a lysis buffer (1% NP-40, 500 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM Benzamide, 1 µl/ml Trypsin inhibitor) containing the Xpert Protease Inhibitor Cocktail Solution and the Xpert Phosphatase Inhibitor Cocktail Solution (GenDEPOT, USA). Lysates were incubated for 20 min at 4℃, centrifuged at 11,000 × g for 20 min at 4℃ and protein concentrations were determined by the BCA protein assay (Thermo Scientific, USA). Protein extracts (50 µg) were boiled for 5 min with SDS sample buffer and then subjected to electrophoresis on 8~15% polyacrylamide gels. Proteins were electroblotted onto nitrocellulose membranes and blocked with 5% skim milk (Becton Dickinson, USA) in tris-buffered saline-0.1% Tween 20 (TBS-T) for 1 hr. Primary antibodies for anti-p62, LC3, ATG7, Beclin-1, cleaved caspase-3, PARP, AMPK, p-AMPK, and β-actin were applied. Blots were subsequently washed three times in TBS-T for 5 min and incubated with specific peroxidase-coupled secondary antibodies (Sigma) for 1 hr. Bound antibodies were visualized using an Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA).
Statistical analysis
Each experiment was repeated three times unless stated otherwise. The data were expressed as means±standard deviation (SD) The statistical significance of the differences between treatments was assessed using one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test. Results were considered statistically significant when the p value was less than 0.05.
RESULTS
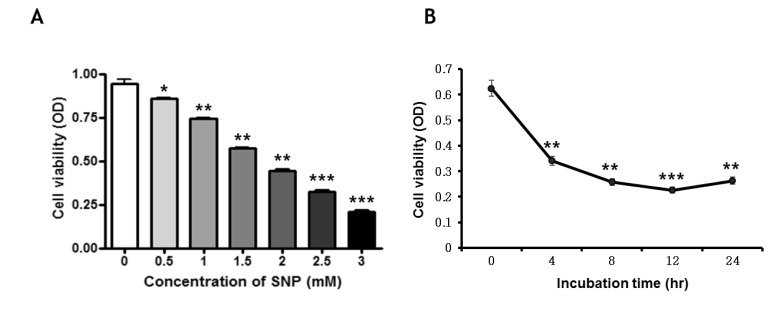
NO decreases cell viability in MC3T3-E1 cells
To examine the effect of NO on the viability of MC3T3-E1 cells, the viability of the SNP-treated cells was measured by MTT assay. Cells were treated with various concentrations (0.5, 1, 1.5, 2, 2.5, and 3 mM) of SNP, an NO donor, for 24 hrs. The viability of MC3T3-E1 cells was reduced by SNP treatment in a dose-dependent manner and it was less than approximately 70% when the cells were treated with 1.5 mM SNP for 24 hrs (Fig. 1A). In addition, treatment of MC3T3-E1 cells with 1.5 mM SNP decreased cell viability in a time-dependent manner (Fig. 1B).
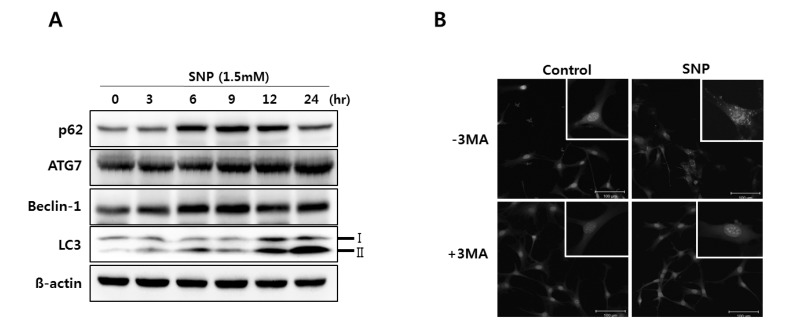
NO induces autophagy in MC3T3-E1 cells
To determine whether NO induces autophagy in MC3T3-E1 cells, the expression of p62, Beclin-1, ATG7 and LC3-II, the markers of autophagy, was determined by western blot analysis in SNP-treated MC3T3-E1 cells. The protein expression of p62, ATG7, and Beclin-1, and the conversion of LC3-I to LC3-II were increased in a time-dependent manner when MC3T3-E1 cells were treated with 1.5 mM SNP for 24 hrs (Fig. 2A). The formation of autophagolysosomes was detected by acridine orange staining. The number of cells with increased acidic autophagolysosomal vacuoles was determined by fluorescence microscopy. The results showed that cells in the control group showed green fluorescence in the nuclei, whereas MC3T3-E1 cells cultured with SNP displayed considerable red fluorescent dots in the cytoplasm, indicating the formation of acidic autophagolysosomal vacuoles (Fig. 2B). In addition, MC3T3-E1 cells were incubated with 1.5 mM SNP without or with 5 mM 3MA, a specific inhibitor of autophagic sequestration, and assessed by fluorescence microscopy following staining with acridine orange. As shown in Fig. 2B, untreated control cells showed considerable red fluorescence, whereas SNP-treated cells incubated with 3MA displayed predominant green fluorescence with very minimal red fluorescence, indicating that the formation of autophagosome as shown in puncta fluorescence (red) can be suppressed by 3MA. These results suggested that NO induces autophagy in MC3T3-E1 cells.
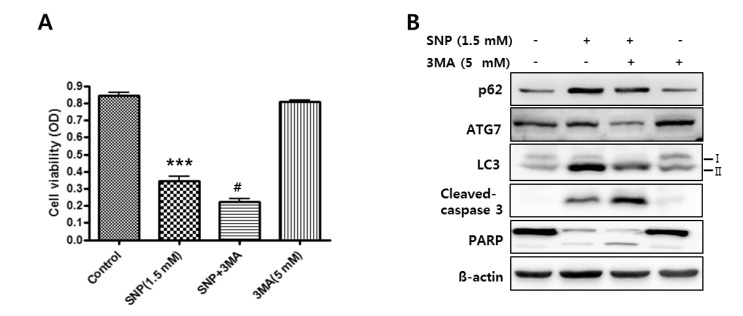
NO-induced autophagy is implicated in cell survival of MC3T3-E1 cells
To determine the role of autophagy in cell death in MC3T3-E1 cells, cells were pretreated with 3MA for 1 hr prior to treatment with 1.5 mM SNP for 24 hrs and MTT assay was performed. 3MA pretreatment significantly decreased cell viability compared to that in cells treated with 1.5 mM SNP only (Fig. 3A). In addition, SNP-treated MC3T3-E1 cells in the presence of 3MA showed decreased LC3 II and p62 expression, whereas they showed increased cleaved caspase-3 and PARP cleavage as the apoptosis markers increased by SNP treatment (Fig. 3B). These results suggested that NO-induced autophagy is associated with cell survival against NO-induced apoptosis of MC3T3-E1 cells.
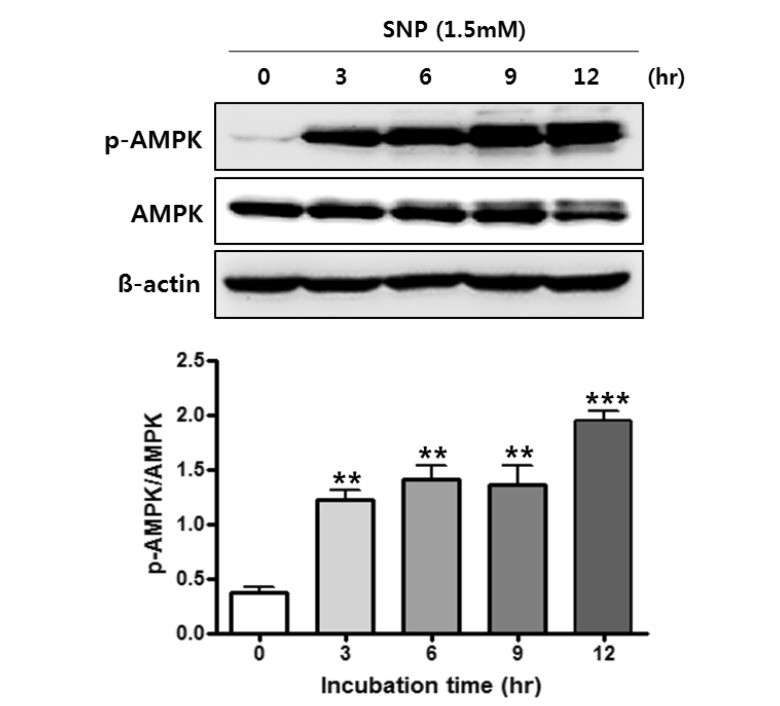
NO activates AMPK in MC3T3-E1 cells
To examine whether NO-induced autophagy in MC3T3-E1 cells activates AMPK signaling, phosphorylation of AMPK was detected by western blot analysis in the SNP-treated MC3T3-E1 cells (Fig. 4). The phosphorylation of AMPK at Thr-172 was increased in a time-dependent manner in the SNP-treated MC3T3-E1 cells. This result suggested that NO activates AMPK, known as an autophagy regulator, in MC3T3-E1 cells.
NO-induced AMPK activation is associated with cell survival in MC3T3-E1 cells
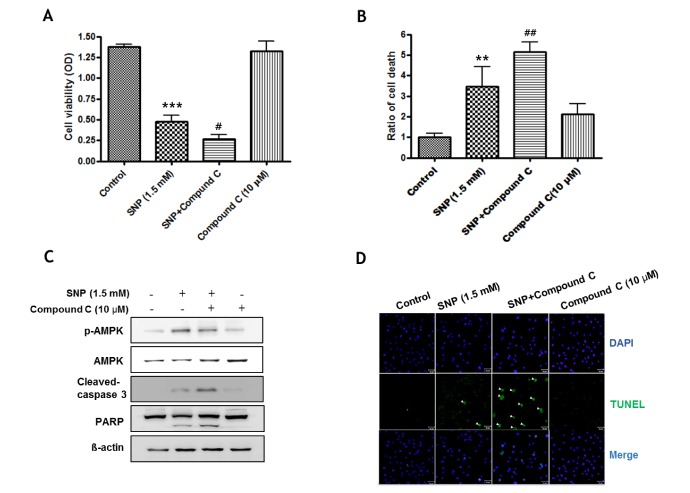
To determine the role of AMPK in NO-induced cell death in MC3T3-E1 cells, MTT assay was performed in cells pretreated with compound C, an inhibitor of AMPK, for 1 hr prior to treatment with 1.5 mM SNP for 24 hrs. Pretreatment with compound C significantly decreased cell viability compared to that in cells treated with 1.5 mM SNP only (Fig. 5A). NO-induced cell death was detected by trypan blue staining. Pretreatment with compound C significantly increased cell death compared to that in cells treated with 1.5 mM SNP only (Fig. 5B). In addition, SNP-treated MC3T3-E1 cells in the presence of compound C showed decreased the phosphorylation of AMPK, whereas they showed enhanced cleaved caspase-3 and PARP cleavage as the apoptosis markers increased by SNP treatment (Fig. 5C). The TUNEL assay detected DNA fragmentation, a hallmark of apoptotic cell death, in MC3T3-E1 cells incubated with 1.5 mM SNP without or with 10 µM compound C. As shown in Fig. 5D, SNP treatment caused the appearance of TUNEL-positive nuclei that showed green fluorescence indicating apoptotic cells, whereas pretreatment of SNP-treated cells with compound C increased the number of cells with TUNEL-positive nuclei compared to that in cells treated with SNP only. These results suggested that NO-induced AMPK activation is associated with cell survival in MC3T3-E1 cells.
DISCUSSION
NO, a reactive free radical containing an unpaired electron is synthesized from L-arginine by NOS, and it is involved in inflammation, immunological function, apoptosis, and autophagy [3637383940]. In a recent report, the inflamed dental pulp cells showed high expression of iNOS which resulted in mass production of NO, and NO-induced autophagy played an important role in survival of inflamed odontoblasts or dental pulp inflammation [414243]. Also, He et al. (2014) demonstrated that physalin A induced both apoptosis and autophagy in human melanoma cells and autophagy plays a protective role against apoptosis by diminishing NO production [44]. It was reported that overproduction of NO by activation of iNOS promotes reduction of osteoblast numbers in bone tissues [1]. However, there are no reports on the role of NO-induced autophagy in MC3T3-E1 cells.
The present study showed that SNP, an NO donor, decreased cell viability of MC3T3-E1 cells in a time- and dose-dependent manner and it increased cleaved caspase-3 and PARP cleavage, suggesting that NO induces apoptotic cell death in osteoblasts, which is consistent with that in previous reports [3]. We investigated whether autophagy occurs during the process of NO-induced apoptosis. SNP treatment increased conversion of LC3-I to LC3-II and expression of p62, ATG7, and Beclin-1, the autophagic markers, in MC3T3-E1 cells. Autophagy is morphologically characterized by an accumulation of autophagosomes, which subsequently fuse with lysosomes to form autophagolysosomes [45]. In the present study, SNP treatment increased acidic autophagolysosomal vacuoles detected by acridine orange staining in MC3T3-E1 cells. These results provided evidence that NO induces autophagy in MC3T3-E1 cells. This finding was consistent with previous reports, indicating that NO can induce apoptosis and autophagy in several cells [41424344].
The role of autophagy in cell survival and cell death is controversial [46]. Several recent studies have reported that autophagy may be cytoprotective against cell death or may induce cell death depending on the type of stimulus and cell type [4447]. In the present study, inhibition of autophagy by 3MA resulted in lower cell viability than that of cells treated with SNP only, whereas it increased cleavage of caspase-3 and PARP in MC3T3-E1 cells, compared to that in cells treated with SNP, demonstrating that co-treatment with the autophagy inhibitor 3MA renders cells more susceptible to NO-induced apoptosis. Based on these results, it can be suggested that NO-induced autophagy is cytoprotective against NO-induced cell death and there is an interplay between NO-induced autophagy and apoptosis in MC3T3-E1 cells.
Inhibition of mTOR (mammalian target of rapamycin) is associated with autophagy induction [48]. AMPK kinase and Akt kinase are two main kinases controlling the mTOR activity. AMPK kinase is involved in inhibition of mTOR activation in response to nutritional, genotoxic, and metabolic stress, which induces autophagy. AMPK plays critical roles in regulating growth, reprogramming metabolism, and cellular processes including cell polarity [4950]. In addition, the Akt kinase mediates the activation of mTOR, which negatively regulates autophagy. Akt is also known to be involved in the regulation of cellular growth, proliferation, and survival and is a principal energy-sensing intracellular enzyme activated in various cellular and environmental stress conditions [51]. We analyzed the activities of AMPK kinase and Akt kinase. The present study showed that AMPK phosphorylation was significantly increased in the SNP-treated MC3T3-E1 cells. However, Akt phosphorylation was unchanged in SNP-treated cells (data not shown). It was consistent with previous report that NO act as an endogenous AMPK activator. This report showed that NO activates AMPK through a guanylyl cyclase-mediated and Ca2+/calmodulin-dependent protein kinase and AMPK activation further increases NO release through AMPK-dependent phosphorylation of endothelial nitric oxide synthase (eNOS) [52].
Furthermore, the role of autophagy in NO-induced cell death of MC3T3-E1 cells via the AMPK pathway was investigated. Co-treatment with compound C, an inhibitor of AMPK, and SNP significantly diminished the cell viability and augmented the number of TUNEL positive cells detected DNA fragmentation as a hallmark of apoptotic cell death, compared to that in cells treated with SNP only. In addition, compound C enhanced cleaved caspase-3 and PARP cleavage, apoptosis markers, increased by SNP treatment. These results showed that NO-induced autophagy was mediated via AMPK activation, and inhibition of AMPK resulted in the increase of apoptosis in NO-treated MC3T3-E1 cells, suggesting that NO-induced AMPK activation plays an important role in defense against NO-induced cell death in MC3T3-E1 cells. This finding is consistent with the previous report, which showed AMPK activation protects against hydrogenperoxide-induced osteolast apoptosis throough autophagy induction and other report, which indicated that enhanced autophagy activity in senescent fibroblasts plays a cytoprotective role during the senescence process leading to AMPK activation [5354]. Meanwhile, a recent study demonstrated that compound c reduced cell viability both by inhibiting proliferation and inducing cell death by multiple mechanisms. The anti-proliferative effects of compound C are mediated by inhibition of AMPK as well as AMPK-independent mechanism, including activation of the calpain/cathepsin pathway, inhibition of Akt, mTOR, cell cycle block, and induction of necroptosis and autophagy [55]. In the present study, it had the possibility that effects of compound C may be AMPK-dependent as well as AMPK-independent, although compound C inhibited AMPK activation.
On the other hand, it was reported that AMPK activated by NO can contribute to the anti-inflammatory actions through inhibiting NF-κB in endothelial cells [56]. Also, AMPK negatively regulates cytokine synthesis such as IL-6 through the IκB/NF-κB pathway in osteoblasts [57]. It have since been reports crosslinking AMPK with NF-κB, but it is controversial as both NF-κB inhibition and activation by regulation of AMPK. Therefore, further study needs whether NF-κB pathway is involved in cytoprotection of NO-induced autophagy mediated via AMPK activation in MC3T3-E1 cells.
Based on these results, it can be concluded that NO induced both apoptosis and autophagy in MC3T3-E1 cells, and autophagy mediated by AMPK activation plays a cytoprotective role against NO-induced cell death.
 XML Download
XML Download