

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
Ang II
angiotensin II
AT1
Angiotensin II type 1 receptor
IGFBP-5
insulin-like growth factor
VSMC
vascular smooth muscle cell
ROS
reactive oxygen species
MAPK
mitogen activated protein kinase
ERK1/2
extracellular signal regulated kinase 1/2
Egr-1
early growth response-1
PI3K
phosphoisositide 3 kinase
SHR
spontaneously hypertensive rats
WKY
wistar kyoto rats
ECL
electrochemiluminescence
MTT
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolidium bromide
SDS
sodium dodecyl sulfate
PVDF
polyvinylidene difluoride
INTRODUCTION
The insulin-like growth factors (IGFs) are normally bound to insulin-like growth factor binding proteins (IGFBPs) [1]. IGFBPs regulate the actions of IGFs in endocrine, paracrine, and autocrine settings [2]. IGFBPs 1~6 evolved into high-affinity IGF binders to influence cell growth by both IGF-dependent and IGFBPs 7~10 into low-affinity IGF binders by IGF-independent mechanisms [2]. IGFBP5, the most conserved IGFBP in all vertebrates [3], is closely related to embryonic development [4], various cancer development [56789] and differentiation of skeletal muscle [10]. In particular, IGFBP5 is associated with cell proliferation [11], cell survival [111213] and interaction with extracellular matrix proteins (ECM) [141516]. In our previous study, IGFBP5 was the only IGFBP5 that was expressed at a higher level in the VSMC from hypertensive compared to those from normotensive rat [17]. Previous studies have shown that IGFBP5 stimulates cell growth in human intestinal smooth muscle by Ras-dependent activation of p38 MAP kinase and extracellular signal-regulated kinase (ERK1/2) pathways [18]. In addition, IGFBP5 plays an important role in regulation of VSMC proliferation via ERK1/2 signaling [17].
Angiotensin II (Ang II), a key mediator of hypertensive, causes structural changes in the arteries vascular remodeling, which involve alterations in cell growth, VSMC hypertrophy, and ECM accumulation [192021]. The physiological actions of Ang II are induced via its 2 main cell surface receptors; AT1 (type 1 receptor), AT2 (type 2 receptor) [2223]. AT1 is upregulated in cardiovascular diseases, including atherosclerosis, hypertension, cardiac hypertrophy, and heart failure [24]. Activation of AT1 induces a cascade of phosphorylations which activate MAPK [25]. Acutely, increased levels of Ang II lead to an increased level of AT1R activation which has been shown to activate signaling cascades that activate MAPKs, including ERK1/2, JNK, and p38, which are implicated in VSMC differentiation, proliferation, migration, and fibrosis [26]. Upregulation of Egr1 was recently shown to be a key contributing factor in stimuli-induced VSMC proliferation and hypertrophy, and Egr1 expression is upregulated in response to Ang II [2728]. Ang II induced EGR1 in VSMC is mediated through the AT1, but not AT2 receptor [29]. Several studies have reported an association of ERK-dependent expression of Egr1 with proliferation and growth in endothelial cells and VSMC [3031]. Furthermore, Ang II stimulates Akt/PKB activity via AT1 receptors in VSMC [32].
Statins are a class of drugs used to lower cholesterol levels by inhibiting the enzyme HMG-CoA reductase, which plays an important role in the production of cholesterol in liver [33]. A recent study demonstrated that Statins have significant immunomodulatory effects and reduce vascular injury [34]. Simvastatin prevented the development of hypertension and cardiovascular hypertrophy together with inhibition of the induced Ang II production of ROS [35]. In particular, Pitavastatin inhibited VSMC proliferation via inactivation of ERK1/2 [36].
In previous studies, VSMC from SHR proliferated more rapidly than those from normotensive WKY [3738]. In addition, IGFBP5 is endogenously up-regulated in VSMC of hypertensive rats [18]. They showed that IGFBP5 stimulates VSMC proliferation in normotensive rat arteries by activating the ERK1/2 MAPK signaling pathway [18]. We therefore wondered not only whether Ang II induced IGFBP5 but also whether IGFBP5 facilitates proliferation and migration in VSMC. However, the signaling mechanism for Ang II induced IGFBP5 effects has not been clearly defined. In the current study, we demonstrate that IGFBP5 might be upregulated by Ang II, and IGFBP5 expression might contribute to proliferation and migration through ERK1/2, p38, Akt, and Egr1 signaling pathways in VSMC. With this background, we hypothesize that Pitavastatin may play a critical role in modulating the action of Ang II-induced IGFBP5 on VSMC proliferation and migration. Thus, we examined which signaling is involved in Ang II-induced IGFBP5 effects and how Pitavastatin modulates the signaling mechanism of Ang II-induced IGFBP5. Our results indicate that Ang II induced IGFBP5 via ERK1/2, p38, Akt, and Egr1. Pitavastatin modulates Ang II induced IGFBP5 expression, cell proliferation, and migration via IGFBP5.
METHODS
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and antibiotics (penicillin and streptomycin) were purchased from Gibco BRL (Rockville, MD, USA). Anti-β-actin, 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-IGFBP5, anti-JNK, anti-pJNK, anti-p38, anti-pp38, anti-ERK1/2, anti-pERK1/2, anti-Akt, goat anti-rabbit IgG, goat anti-mouse IgG, donkey anti-goat IgG, enhanced chemiluminescence (ECL), IGFBP5, Egr1, and control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Ang II, PD98059, U0126, SP600125, SB203580, and LY294002 were purchased from Calbiochem (San Diego, CA, USA). ERK1/2 and control siRNA were purchased from Thermo Scientific Dharmacon (Chicago, IL, USA), Lipofectamine2000™ was purchased from Invitrogen (Carlsbad, CA, USA), recombinant IGFBP5 was purchased from R&D system (Minneapolis, ME, USA).
Cell Culture
A7r5 smooth muscle cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained under 5% CO2 at 37℃ in Dulbecco's modified Eagle's medium with l-glutamine and 4,500 mg/L glucose containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA). They were sub-cultured every 3 or 4 days with trypsin (0.25%) and EDTA (0.02%). A portion of the detached cells were transferred to normal Tyrode's (NT) solution and kept at 4℃ in micro-tubes.
Western blot analysis
Whole cell extracts were prepared by lysing cells in RIPA buffer protein extraction solution. Protein concentrations were quantified with protein assay reagent (Bio-Rad, Hercules, CA, USA). Equal amounts of protein were mixed with sodium dodecyl sulfate (SDS) sample buffer and incubated for 5 min at 100℃ before loading. Total protein samples (30 µg) were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) for 1 hr and 30 min at 100 V. The separated proteins were then transferred onto PVDF membranes for 1 hr at 30 mA using a SD Semi-dry Transfer Cell. The membranes were blocked with 5% non-fat milk in PBS-T (phosphate buffered saline containing 0.05% Tween 20) for 1 hr at room temperature, followed by incubation with the primary antibodies at a dilution of 1:1,000 in 5% skim milk in PBS (phosphate buffered saline) overnight at 4℃. The membranes were then washed with PBS-T, followed by incubation with the secondary antibodies in 5% skim milk in PBS for 1 hr at room temperature. Finally, after rinses with the wash buffer, the membranes were exposed to ECL western blot analysis detection reagents (MicroChemi 4.2 Chemilumineszenz-System, Israel).
Cell proliferation assay
Cell numbers were determined after 24 or 48 hours of stimulation with 10% FBS using a hemocytometer, and cell viability was determined using the MTT assay. VSMC were seeded onto 24-well plates at a density of 1×104 cells per well in DMEM supplemented with 10% FBS. After different treatments, 50 µl of 1 mg/ml MTT solution was added to each well (0.1 mg/well), followed by incubation for 4 hrs. The supernatants were then aspirated, and the formazan crystals were solubilized with 200 µl of dimethyl sulfoxide (DMSO). The supernatants (100 µl) of these solutions were placed in the wells of 96-well plates. Cell proliferation was determined using a microplate reader (Bio-Rad, Hercules, CA, USA) measuring absorbance at 570 nm.
Migration assay
Cell migration was analyzed by a scratch wound assay. Cells were grown to confluence in 6-well plates. A scratch wound was made in the monolayer by dragging a yellow pipette tip across the layer. Cells were washed away with PBS. The cells were cultured with Ang II or without Ang II for 24 hrs. Cells were photographed immediately and 24 hrs after the scratch with a Nikon digital camera. The wound area was then measured to determine cell migration.
Transfection of siRNA
The cells were transfected with siRNA using Lipofectamine 2,000 reagent, according to the manufacturer's instructions. Aliquots of 1×104 cells were plated on 60-mm dishes 1 day before transfection and grown to approximately 70% confluence. The cells were then transfected with siRNA (20 nM IGFBP5, 20 nM control) and 3 µl of lipofectamine for 6 hrs in Opti-MEM®I reduced serum medium (Invitrogen, Carlsbad, CA, USA), followed by incubation for 48 hrs. Protein levels were determined by Western blot and cell proliferation using an MTT assay.
RESULTS
IGFBP5 expression is induced by Ang II in VSMC
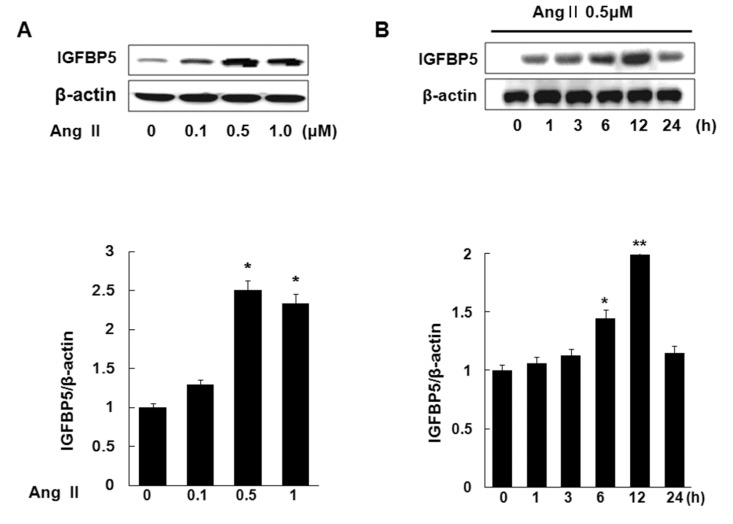
IGFBP5 is upregulated in both VSMC of hypertensive rats and Ang II-stimulated VSMC [18]. To determine whether Ang II induces IGFBP5 protein level in VSMC, cells were treated with Ang II 0, 0.1, 0.5, 1.0 µM respectively for 24 hrs. Ang II increased IGFBP5 expression in a dose-dependent manner. IGFBP5 expression peaked at 0.5 µM Ang II. The special expression of IGFBP5 at 0.5 µM showed an approximate 2.5 fold increase over the control (p<0.05) (Fig. 1A). Cells were treated with 0.5 µM Ang II at various times, 0, 1, 3, 6, 12, and 24 hrs. Ang II increased IGFBP5 expression in a time-dependent manner. IGFBP5 expression showed a dramatic increase at 12 hrs (Fig. 1B). These results suggest that Ang II induces IGFBP5 in time- and dose-dependent manner in VSMC.
Pitavastatin reduces induction of pERK1/2, p38, pAkt, and Egr1 expression by Ang II in VSMC
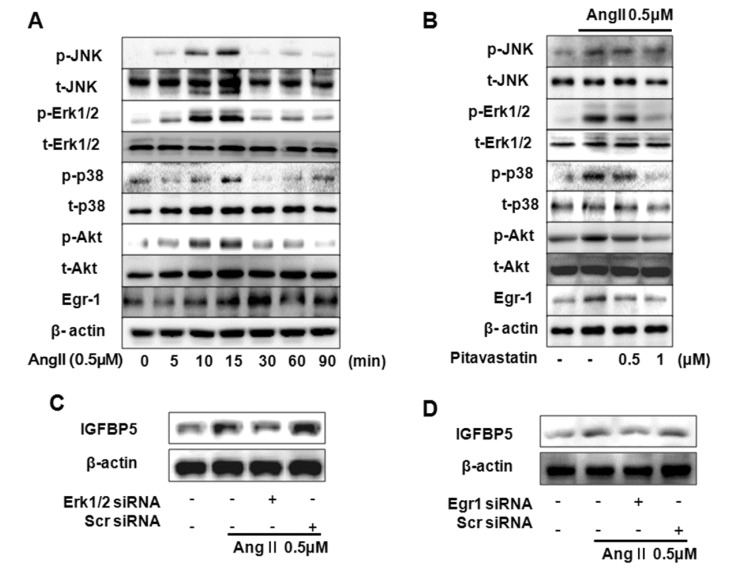
Upregulation of Ang II has been shown to activate signaling cascades that activate MAPKs (ERK1/2, JNK, p38), Akt, and Egr1 [262932]. We demonstrate activation of MAPKs, Akt and Egr1 by Ang II. Ang II induced p-ERK1/2, p-P38, p-JNK, p-Akt, and Egr1 after 10 min with 0.5 µM Ang II stimulation. p-MAPKs and p-Akt peaked after 15 min and Egr1 peaked after 30 min (Fig. 2A). To evaluate the effect of Pitavastatin on Ang II induced MAPKs, Akt and Egr1 activation, cells were treated with Pitavastatin 0.5 µM and 1.0 µM, respectively. Pitavastatin (1.0 µM) inhibited Ang II induced activation of ERK1/2, p38, Akt, and Egr1 in VSMC. However JNK was not induced by Ang II in VSMC (Fig. 2B). In addition, we confirmed that Pitavastatin had no inhibition effect on Ang II-activated AT1R (data not shown). Knockdown of ERK1/2 resulted in complete inhibition of Ang II induced IGFBP5 expression in VSMC (Fig. 2C). Knock down with Egr1 resulted in complete inhibition of Ang II induced expression of IGFBP5 in VSMC (Fig. 2D). These results suggest that Pitavastatin modulates Ang II-induced ERK1/2, p38, (but not JNK), PI3K, and Egr1.
Ang II induces VSMC proliferation via AT1-dependent IGFBP5 induction
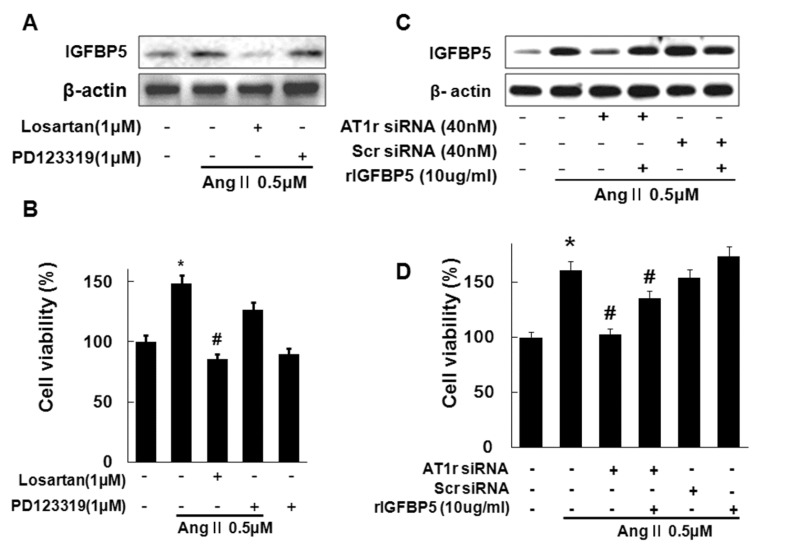
In our previous figure, we confirmed that Ang II induced IGFBP5 in VSMC. However, whether AT1 or AT2 receptors activate IGFBP5 has not been clearly defined. Therefore we first examined whether AT1 or AT2 receptors activate IGFBP5. Ang II-induced IGFBP5 expression was blocked by AT1R antagonist, Losartan, but not AT2R antagonist, PD123319 (Fig. 3A). In addition, Losartan inhibited Ang II-induced cell proliferation in VSMC (Fig. 3B). Then knockdown of AT1R resulted in significantly inhibited Ang II-induced expression of IGFBP5 in VSMC. IGFBP5 recombinant protein reversed inhibition of IGFBP5 expression by AT1R siRNA in VSMC (Fig. 3C). In addition, knockdown with AT1R resulted in significantly inhibited Ang II-induced cell proliferation. IGFBP5 recombinant reversed inhibition of Ang II-induced VSMC cell proliferation by AT1 siRNA (Fig. 3D). Together, these findings suggest that Ang II activates IGFBP5 via AT1 receptor and Ang II induced VSMC proliferation via induction of IGFBP5.
Ang II-induced IGFBP5 expression through ERK1/2, p38, and Akt
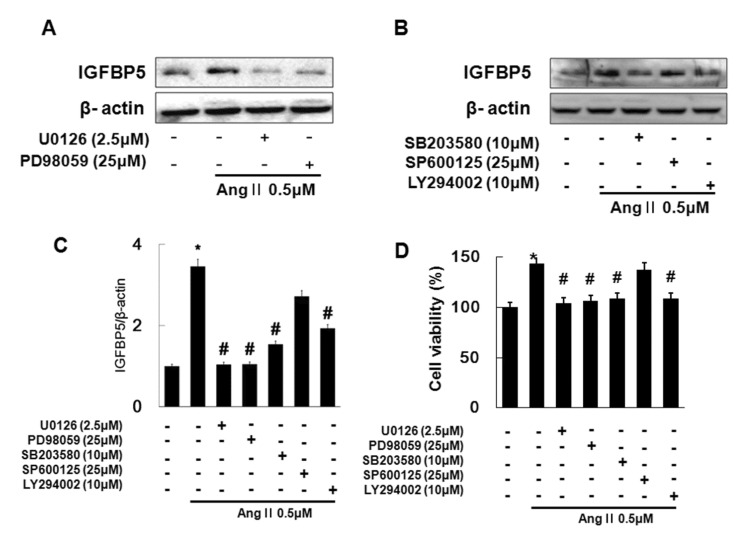
In the previous study, Ang II activated ERK1/2, p38 and JNK, PI3K and Egr1 (Fig. 2). We investigated which signaling was involved in Ang II-induced IGFBP5 in VSMC. We first confirmed whether Ang II-induced IGFBP5 is induced by ERK1/2. We treated with MEK/ERK1/2 inhibitors such as U0126 (1 µM), PD98059 (1 µM) for 45 min, followed by treatment with Ang II for 12 hrs in VSMC. Next, we investigated whether Ang II-induced IGFBP5 is induced by p38, JNK, and PI3K. We treated VSMC with p38 inhibitor; SB203580 (10 µM), JNK inhibitor; SP600125 (25 µM) and PI3K inhibitor; LY294002 (10 µM) for 45 min, followed by treatment with Ang II for 12 hrs. IGFBP5 was inhibited by the MEK1/2 inhibitors in Ang II treated VSMC (Fig. 4A). And also IGFBP5 expression was significantly inhibited by the p38 inhibitor and PI3K inhibitor in Ang II treated VSMC. However, treatment with JNK inhibitor had no inhibitory effect in Ang II treated VSMC (Fig. 4B and 4C). In addition, Ang II-induced cell proliferation was inhibited by the MEK1/2, p38 and PI3K inhibitors not JNK inhibitor in VSMC (Fig. 4D). These results demonstrate that Ang II induced IGFBP5 and cell proliferation via ERK1/2, p38, and PI3K signaling in VSMC.
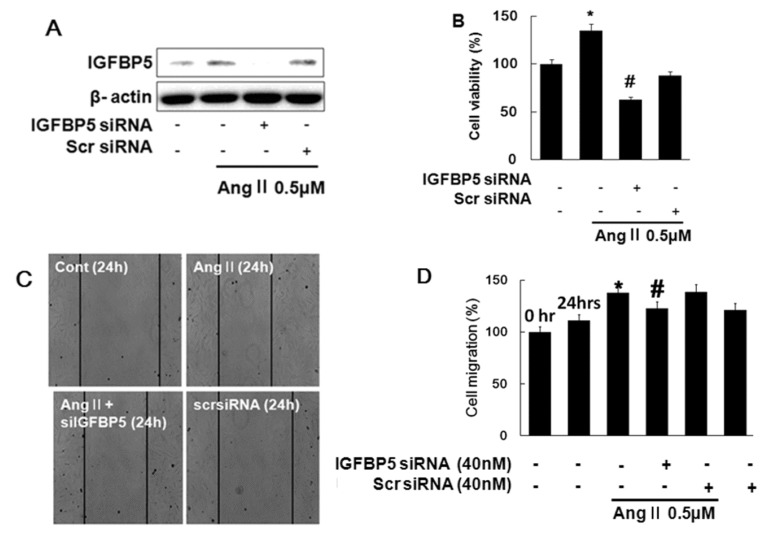
IGFBP5 knockdown modulates Ang II-induced proliferation and migration
To determine whether the IGFBP5 expression is due to Ang II stimulation, cells were transfected with IGFBP5 siRNA. Knockdown of IGFBP5 suppressed IGFBP5 expression (Fig. 5A). Knockdown of IGFBP5 resulted in inhibition of Ang II-induced cell proliferation in VSMC (Fig. 5B). Knockdown of IGFBP5 resulted in suppression of Ang II-induced cell migration in VSMC (Fig. 5C). Migration was estimated by measuring the cell area within the wound region (Fig. 5D). These results indicated that IGFBP5 knockdown modulates Ang II induced proliferation and migration via IGFBP5 induction.
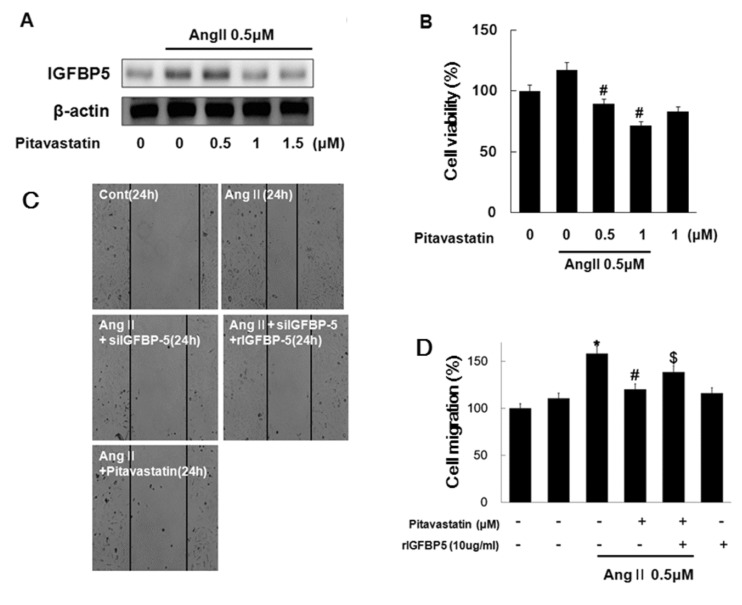
Pitavastatin regulates Ang II-induced IGFBP5 expression, proliferation and migration
Pitavastatin inhibited VSMC proliferation via inactivation of ERK1/2 [36]. We performed testing to determine whether Pitavastatin modulates Ang II-induced IGFBP5 expression and other effects such as proliferation and migration. Cells were treated with Pitavastatin 0.5 µM, 1.0 µM, and 1.5 µM, respectively, for 45 min, followed by treatment with Ang II for 12 hrs. Pitavastatin significantly reduced Ang II-induced IGFBP5 protein expression (Fig. 6A). Cells were treated with Pitavastatin 0.5 µM and 1.0 µM for 45 min, followed by treatment with Ang II for 24 hrs. Pitavastatin significantly reduced Ang II-induced IGFBP5 cell proliferation (Fig. 6B). Next, we examined the question of Pitavastatin modulates Ang II-induced VSMC migration via IGFBP5. Pitavastatin significantly inhibited Ang II-induced cell migration in VSMC. In particular, IGFBP5 recombinant reversed modulation of Ang II-induced VSMC migration by Pitavastatin (Fig. 6C and 6D). Our findings suggest that Pitavastatin modulates Ang II induced proliferation and migration in VSMC.
DISCUSSION
Angiotensin II (Ang II) plays a fundamental role in controlling blood pressure and tissue remodeling. Some studies have shown that Ang II plays critical roles in cell proliferation and angiogenesis of various tumors [192021]. VSMC proliferation contributes to the arterial remodeling associated with hypertension and atherosclerosis [20]. Atherosclerosis progression is closely correlated with VSMC proliferation and migration, which requires a coordinated rearrangement of the VSMC. Ang II leads to AT1R activation, which has been shown to activate signaling cascades that activate MAPKs, including ERK1/2, JNK, and p38, which are implicated in VSMC differentiation, proliferation, migration, and fibrosis [22232526]. In addition, upregulation of Egr-1 was recently shown to be a key contributing factor in stimuli-induced VSMC proliferation [2728]. Furthermore, Ang II stimulates Akt/PKB activity via AT1 receptors in VSMC [32]. AT1 is upregulated in cardiovascular diseases including atherosclerosis [24]. We confirmed that AT1R are proposed to be Ang II responsive mediators that can cause IGFBP5 expression, leading to cell proliferation. As shown in Fig. 3, Ang II-induced IGFBP5 expression was blocked by AT1R antagonist, Losartan, but not AT2R antagonist, PD123319 (Fig. 3A). In addition, Losartan inhibited Ang II-induced cell proliferation in VSMC (Fig. 3B). Then knockdown with AT1R resulted in significantly inhibited Ang II-induced cell proliferation and IGFBP5 recombinant reversed inhibition of Ang II-induced VSMC cell proliferation by AT1 siRNA (Fig. 3D). Ang II activates IGFBP5 via AT1 receptor results in VSMC proliferation.
Several studies have reported that IGFBP5 promoted proliferation and migration in VSMC [11121314151639]. IGFBP5 also plays a role in modulation of VSMC proliferation in hypertensive rats via ERK1/2 MAPK signaling [18]. We hypothesize that modulation of Ang II-induced IGFBP5 effects may play an important inhibitory role in cardiovascular diseases including atherosclerosis. We confirmed that IGFBP5 is inhibited by the MEK1/2 inhibitors in Ang II treated VSMC (Fig. 4A). And also IGFBP5 expression was significantly inhibited by the p38 inhibitor and PI3K inhibitor in Ang II treated VSMC. However, treatment with JNK in hibitor had no inhibitory effect in Ang II treated VSMC (Fig. 4B and 4C). In addition, Ang II-induced cell proliferation was inhibited by the MEK1/2, p38 and PI3K inhibitors not JNK inhibitor in VSMC (Fig. 4D). Our results not only confirm that Ang II induced IGFBP5 expression via multiple signaling kinases, but also provide evidence indicating that Ang II-induced IGFBP5 promoted cell proliferation and migration in VSMC. Our results provide new insights into the mechanism underlying Ang II-induced IGFBP5 effects, such as migration and proliferation (Fig. 5).
In several studies, Statins have significant immunomodulatory effects and reduce vascular injury as 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitors [34]. In the current study, Pitavastatin inhibited Ang II-induced proliferation by inactivating ERK1/2 in VSMC [36]. As one of our important objectives, we examined whether Pitavastatin modulates Ang II induced IGFBP5 expression, proliferation, and migration in VSMC. Our results demonstrated that Pitavastatin inhibits activation of ERK1/2, P38, PI3K, and Egr1 by Ang II. This is the first study to report that Pitavastatin has a modulatory effect on Ang II-induced IGFBP5 in VSMC. A previous study showed that Statins have significant immunomodulatory effects and reduce cell proliferation [34]. Therefore, we wondered whether Pitavastatin can inhibit Ang II-induced proliferation and migration in VSMC via IGFBP5. As shown in Figure 6, Pitavastatin significantly inhibited Ang II-induced cell proliferation and migration in VSMC. In particular, IGFBP5 recombinant reversed modulation of Ang II-induced VSMC migration by Pitavastatin. Pitavastatin modulated Ang IIinduced proliferation and migration in VSMC via IGFBP5 expression.
In summary, these findings suggested that Pitavastatin inhibited Ang II-induced expression of IGFBP5 through ERK1/2, p38, and PI3K signaling pathways. IGFBP5 induces VSMC proliferation and migration, which contributes to the progression of atherosclerosis. Taken together, our findings provide evidence that Pitavastatin may play a critical role in Ang II-induced IGFBP5 mediated VSMC formation.
 XML Download
XML Download