

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
AQP4
aquaporin-4
DW
dry weight
EGCG
epigallocatechin-3-gallate
HRP
horseradish peroxidase
GFAP
glial fibrillary acidic protein
GSH-PX
glutathione peroxidase
GTPase
guanosine triphosphatase
NOX2
NADPH oxidase 2
iNOS
nitric oxide synthase
MDA
malondialdehyde
PBS
phosphate-buffered saline
Q-PCR
quantitative real-time polymerase chain reaction
SOD
superoxide dismutase
TBI
traumatic brain injury
WW
wet weight
INTRODUCTION
Traumatic brain injury (TBI) is the most common cause of death and disability among young adults [1]. The damage to the brain occurs in two phases, the initial primary phase is characterized by direct cerebral tissue damage which leads to accumulation of lactic acid, membrane perme-ability increasing and consecutive edema formation [2]. The secondary phase is characterized by a variety of physiological, cellular, and molecular responses aimed at restoring the homeostasis of the damaged tissue [3]. These events trigger cellular structures damages, vascular membrane degradation and necrotic or programmed cell death [4]. However, the increasing clinical analyses and experiments of biomechanical injury have expanded the knowledge of pathophysiological events and represented a window of opportunity in which pharmaceutical compounds with neuroprotective properties could be administered [15].
Numerous evidences have shown that oxidative stress plays a key role in the development of TBI [6]. The consecutive Ca2+ and Na+ influx, which lead to catabolic intracellular processes, is an early event that occurs within minutes of mechanical impact. Ca2+ activates lipid peroxidases, proteases and phospholipases which in turn increase the intracellular concentration of free fatty acids and free radicals including superoxide and hydroxyl radicals [12]. It has been reported that the inhibition of NADPH oxidase activation, which contribute to cellular ROS production and cause oxidative damage of DNA, proteins and lipid to reduce traumatic brain injury-induced neuronal death [7]. Free radical scavenging may have a high therapeutic potential for traumatic brain injury.
Epigallocatechin-3-gallate (EGCG) is the main polyphenol extracted from green tea, which is thought to be responsible for the majority of biological activity of green tea [8]. Numerous studies have identified multiple functions for EGCG including anti-inflammation [910], anti-mutagenic [11], anti-infective [12] and anti-cancer properties [1314]. Moreover, EGCG has been demonstrated to regulate signal transduction pathways including JAK/STAT, MAPK, PI3K/AKT, Wnt, Notch, NF-κB and AP-1 [15]. As an antioxidant, EGCG displays a selective anti-apoptotic effect against inducers of mitochondrial oxidative stress in neurons [16]. EGCG also attenuates oxidative stress-induced human neuroblastoma SH-SY5Y cell death through up-regulation of PKC phosphorylation and modulation of cell survival/cell cycle genes [17].
Recent studies indicated that EGCG had neuroprotective effects against neuronal damage and brain edema [1819]. However, the effect of EGCG on traumatic brain injury remains unknown. In this study, a weight drop model was established to evaluate the therapeutic potential of EGCG on TBI and its underlying mechanism.
METHODS
Animals
Male Sprague-Dawley rats (6~8 weeks old, 220~250 g) were purchased from Beijing HFK Bio-Technology Co., Ltd (Beijing, China). They were maintained with free access to pellet food and water in plastic cages at 21±2℃ and kept on a 12 h light/dark cycle. Animal welfare and experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006) and the related ethical regulations of Tianjin Medical University. All the animal experiments were approved by Tianjin Medical University Animal Care and Use Committee.
Establishment of TBI model
Feeney's weigh-dropping model was established to assess the effect of EGCG on TBI as previously described [20]. Briefly, after anesthesia with pentobarbital sodium (50 mg/kg), rats were fixed in a stereotaxic frame and the scalp was shaved. A midline incision was made to expose the skull from the bregma to the lambda sutures. The left lateral aspect of the skull was exposed by retracting the skin and surrounding soft tissue. Focal brain trauma was induced by dropping a 20 g steel weight with a flat end from a height of 30 cm onto the left lateral skull. Then the scalp was sutured. After impact, the rats were monitored during recovery of spontaneous respiration. Sham-injured control animals underwent anesthesia and scalp incision, but did not undergo trauma.
Experimental groups
EGCG (Sigma-Aldrich Co, St Louis, MO) was dissolved in phosphate-buffered saline (PBS) to a final concentration of 20 mg/ml. All rats were randomly assigned to the following groups: sham+vehicle (PBS), TBI+vehicle (PBS) and TBI+EGCG. After TBI, rats were administered immediately with PBS or 100 mg/kg EGCG intraperitoneally according to previous study [18]. At 12, 24, 48 and 72 h following trauma, rats were sacrificed for analysis.
Evaluation of brain edema
To evaluate brain water content, a sensitive measure of cerebral edema was determined using the wet weight (WW) to dry weight (DW) ratio method, as previously described [18]. In brief, after TBI, rats were sacrificed at indicated time, and their left brain hemispheres were rapidly removed and weighed to obtain WW. The tissue was dried in 70℃ oven for 72 h and weighed again to obtain DW. The percentage of tissue water content was calculated as (WW-DW)/WW×100%.
Measurement of SOD, GSH-PX and MDA activity
The levels of oxidative stress-superoxide dismutase (SOD), glutathione peroxidase (GSH-PX) and malondialdehyde (MDA) were measured using a commercially available kit (Nanjing Jiancheng bioengineering institute, Nanjing, China). Briefly, after TBI, the left brain hemispheres of mice were removed and homogenized in ice-PBS. The samples were centrifuged at 2000 g for 10 minutes at 4℃. The supernatant was used for the measurement of SOD, GSH-PX and MDA activities according to the manufacturers' instructions.
Evans blue dye analysis
Extravasations of evans blue dye (Sigma Chemical, USA) into the interstitium was used as a quantitative measure of changes of microvascular permeability in traumatic brain injury [21]. Evans blue dye (0.2 ml/100 g) was injected through the femoral vein. After 2 hours following Evan blue dye injection, rats were sacrificed and the brains perfused with 0.9% saline via the left ventricle. The brain tissue was weighed and homogenized in formamide and incubated at 60℃ overnight. The supernatant was collected by centrifugation at 4,000 g for 30 minutes. Evans blue dye in the brain tissue was quantitated by spectrophotometric analysis at 620 nm. Sample values were compared with those of evans blue dye standards mixed with the solvent (100~1000 ng/ml). Results were expressed in micrograms of evans blue dye per gram of tissue.
Quantitative real-time polymerase chain reaction (Q-PCR)
Real-time PCR was performed as described previously [22]. Briefly, the brain tissues were homogenized in Trizol reagent (Life Technologies, Carlsbad, CA) and total RNA was extracted following the manufacturer's instructions. 1 µg total RNA from each sample was reverse transcribed into cDNA using a RETROscript® reverse transcription kit (Life Technologies, Carlsbad, CA). Quantitative PCR was performed with ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA) using SYBR® Green PCR Master Mix (Life Technologies, Carlsbad, CA). Conditions for amplification were 1 cycle of 94℃ for 5 min followed by 40 cycles of 94℃ for 30 s, 58℃ for 30 s, and 72℃ for 30 s. The primer sequences used in this study were as follows: CD68 F: 5'-CTGTTGCGGAAATA CAAGCA-3', R: 5'-GGCAGCAAGAGAGATTGGTC-3'; IL-1β F: 5'-GACCTGTTCTTTG AGGCTGACA-3', R: 5'-CTCATCTGGACAGCCCAAGTC-3'; TNF-α F: 5'-CATCTTCT CAAAACTCGAGTGACAA-3', R: 5'-TGGGAGTAGATAAGGTACAGCCC-3'; GAPDH F: 5'-ATGACTCTACCCACGGCAAG-3', R: 5'-CTGGAAGATGGTGATGGGTT-3'.
Western blot analysis
Briefly, the left brain hemispheres of mice were removed and homogenized in RIPA buffer (Cell Signaling Technology, Danvers, MA). After 12,000 g centrifugation for 10 min, the protein content of the supernatant was determined by a BCA™ protein assay Kit (Pierce, Rochford, IL). The protein lysates were separated by 10% SDS-PAGE and subsequently electro-transferred onto a polyvinylidene diuoride membrane (Millipore Corp.,Bedford, MA). The membrane was blocked with 5% nonfat milk for 1 hour at room temperature. The blocked membrane was incubated with AQP4, GFAP and GAPDH antibodies (1:1000; Abcam, Cambridge, MA) overnight at 4℃. The next day, the membranes were blotted with horseradish peroxidase (HRP)-conjugated secondary antibody (1:1000; Santa Cruz biotechnology, Santa Cruz, CA) for 1 h. Protein bands were visualized using Western blotting detection system according to the manufacturer's instructions. The densities of protein bands were determined by Image J software.
Membrane proteins were prepared with a Mem-PER Plus Membrane Protein Extraction Kit (Thermo, USA) according to the manufacturer's instructions. Briefly, the tissue were homogenized in the permeabilization buffer using Dounce tissue grinder and incubated at 4℃ for 10 minutes. Cytosolic proteins were separated by centrifugation at 16,000 g for 15 minutes at 4℃. The pellet was resuspended in solubilization buffer and incubated at 4℃ for 30 minutes. Membrane and membrane-associated proteins were collected by centrifugation at 16,000 g for 15 minutes at 4℃. The anti-p47phox, anti-rac1, anti-gp91phox antibodies were purchased from Santa Cruz Biotechnology, Inc.
Immunohistochemistry
After TBI, the left brain hemispheres of mice were kept in 10% formalin solution and paraffin-embedded sections (thickness: 5 µM) were prepared subsequently. After deparaffinization, antigen unmasking was performed in sub-boiling sodium citrate buffer for 10 minutes. Then sections were quenched for 10 minutes in 0.3% hydrogen peroxide and incubated with anti-AQP4 or anti-GFAP antibody (1:100; Abcam, Cambridge, MA) overnight at 4℃. After washing with PBS, the sections were incubated with biotinylated secondary antibodies at room temperature for 1 hour and immunostained by using an avidin-biotin comprex staining kit (Santa cruz biotechnology) according to the manufacturer's instructions. The DAB-positive areas in 5 regions were measured using microscope (OLYMPUS, Japan) and The immuno-histochemical score was calculated by combining an estimate of the percentage of immunoreactive cells (quantity score) with an estimate of the staining intensity (staining intensity score) as previously described [23], as follows: no staining is scored as 0, 1~10% of cells stained scored as 1, 11~50% as 2, 51~80% as 3, and 81~100% as 4. Staining intensity is rated on a scale of 0 to 3, with 0=negative; 1=weak; 2=moderate, and 3=strong.
Statistical analysis
All data were expressed as the mean±standard error of measurement (S.E.M.). Student's t-test was used for statistical analyses of the data. All statistical analyses were conducted using SPSS 10.0 statistical software (SPSS). p-values less than 0.05 were considered to be statistically significant.
RESULTS
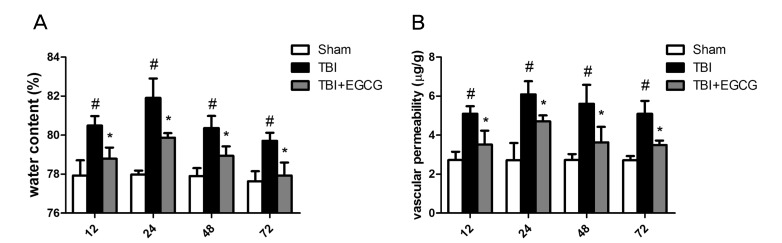
EGCG reduced TBI-induced brain tissue water content and vascular permeability
Edema formation, measured as an increase in brain tissue water content, was analyzed in the peri-contusion area at 12, 24, 48 and 72 h after TBI. As shown in Fig. 1A, the brain tissue water content was significantly increased in TBI group compared with sham group after injury. However, the brain tissue water content after injury was significantly reduced in 100 mg/kg EGCG treatment group compared with TBI group at 12, 24, 48 and 72 h. Earlier studies have established that TBI results in an increasing of vascular permeability [24]. To assess the effect of EGCG on TBI-induced vascular permeability, evans blue dye in the brain tissue was quantified. As shown in Fig. 1B, vascular perme-ability was significantly increased in TBI group compared with sham group at 12, 24, 48 and 72 h after injury. In EGCG group, the extents of vascular permeability were significantly down-regulated in 12, 24, 48 and 72 h after injury compared with TBI group.
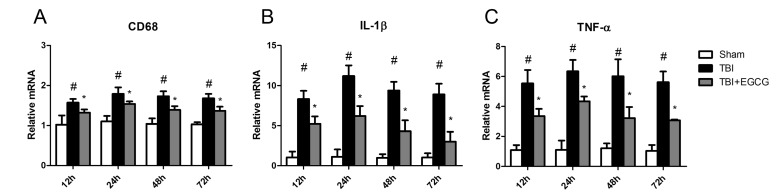
EGCG ameliorated TBI-induced inflammation
Inflammation is an stereotypical response to tissue damage and has been extensively documented in experimental and clinical TBI. Microglia orchestrates many aspects of this response [25]. We further examined the effect of EGCG on TBI-induced inflammation. As shown in Fig. 2, the level of CD68 mRNA expression, as a marker of microglia, was increasing after TBI. EGCG significantly reduced CD68 mRNA expression in the brain. EGCG also reduced TNF-α and IL-1β mRNA expression, suggesting that EGCG inhibited TBI-induced microglia activation.
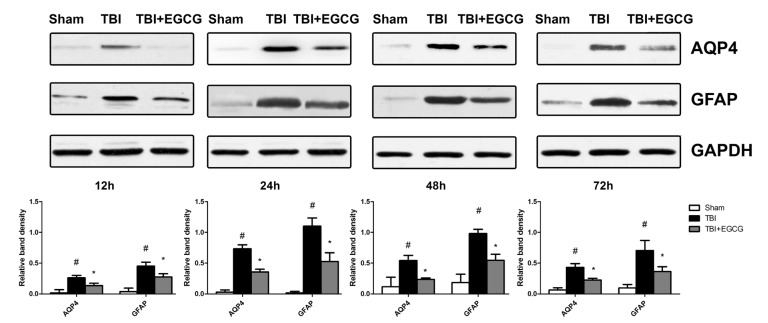
EGCG inhibited AQP4 and GFAP expression
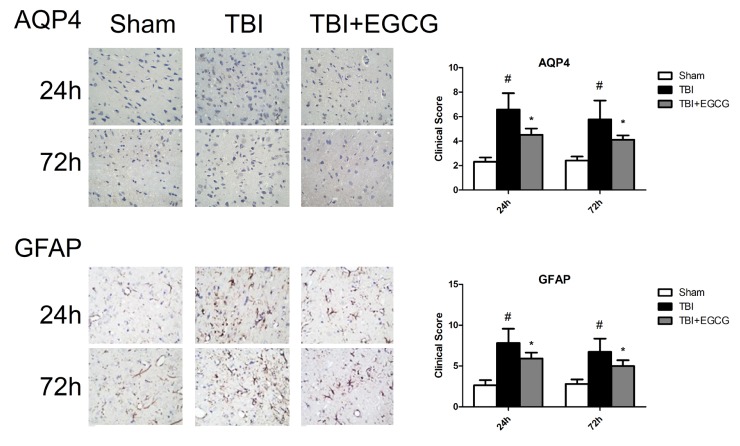
Aquaporin 4, also known as AQP4, plays a significant role in maintaining brain water homeostasis [26]. Western blot result showed that AQP4 expression was at low level in sham group and significantly increased after TBI for 12, 24, 48 and 72 h. Administering of EGCG significantly decreased the level of AQP4 compared with TBI group after injury. Glial fibrillary acidic protein (GFAP) as a biomarker has been used to assess brain damage following TBI [27]. As shown in Fig. 3, EGCG significantly decreased the level of GFAP expression after TBI. Immunohistochemistry results showedn that clinical scores were significantly increasing in TBI group compared with sham group at 24 and 72 h. EGCG treatment significantly down-regulated clinical scores after TBI at 24 and 72 h (Fig. 4).
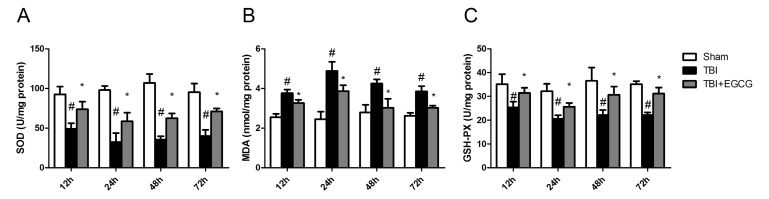
EGCG inhibited oxidative stress after TBI
To assess the effects of EGCG on TBI-induced oxidative stress, the levels of SOD,GSH-PX and MDA were detected after TBI. As shown in Fig. 5, TBI significantly decreased the tissue SOD and GSH-PX activities compared with the sham group. The EGCG administration showed a protective effect against TBI by markedly increasing the antioxidant enzyme SOD and GSH-PX activities and decreasing MDA activity compared with the TBI group.
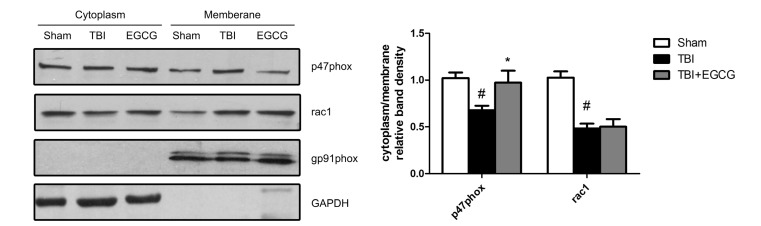
EGCG inhibited TBI-induced p47phox translocation from cytoplasm to plasma membrane
NADPH oxidases are the main enzymes that produce ROS. gp91phox and p47phox are the key constructive components of NADPH oxidase 2 (NOX2), which is an NADPH oxidase that is primarily expressed in microglia and the spinal cord [28]. Western blot analysis showed that p47phox subunit translocated to the neuronal plasma membrane at 24 hour after TBI, which was reduced by EGCG. However, EGCG did not affect translocation of rac1 and the level of gp91phox expression (Fig. 6). The data indicated that EGCG decreased NADPH oxidase activation through inhibition of p47phox translocation.
DISCUSSION
In this study, we investigated the effect of EGCG, the most biologically active compound in green tea, on traumatic brain injury and its underlying mechanism. Our results showed that EGCG significantly inhibited TBI-induced edema formation and decreased vascular permeability. TBI-induced inflammation was reduced by EGCG treatment. Moreover, EGCG treatment significantly inhibited AQP4 and GFAP expression in injured brain tissue. As an anti-oxidant, EGCG ameliorated oxidative stress after TBI via inhibition of p47phox translocation from cytoplasm to plasma membrane. Therefore, our data revealed that EGCG could be therapeutically effective in modulating the pathological process after TBI.
The specific pathophysiology of traumatic brain injury involves cerebral metabolic dysfunction, excitotoxicity, oxidative stress, edema formation, inflammation and cell death (necrosis and apoptosis) [4]. Although a number of factors contribute to the high mortality and morbidity associated with TBI, the development of cerebral edema with brain swelling remains the most significant predictor of outcome. Edema formation lead to an expansion of brain volume as it increases intracranial pressure, impairs cerebral perfusion and oxygenation, and additionally contributes to ischemic injuries [15]. Attenuating blood-brain barrier permeability has become a promising approach to managing cerebral edema and associated swelling given that such increases in cranial water content can only be derived from the vasculature [224]. Numerous studies reported that green tea alleviated early BBB damage and acute stress in the brain [2930]. Importantly, the animal study has been shown that EGCG freely passes the blood-brain barrier and reaches the brain parenchyma [31]. Our data showed that administering of 100 mg/kg EGCG significantly decreased TBI-induced cranial water content and also attenuated TBI-induced vascular permeability. Following TBI, various mediators such as glutamate, lactate, arachidonic acid and its metabolites, histamine, free radicals are released which promote vasogenic and/or cytotoxic cerebral edema formation [5]. It has been reported that protection of motor neuron by EGCG is associated with regulating glutamate level in organotypic culture of rat spinal cord [32]. Further detailed research should be able to show whether EGCG could regulate these mediators balance after TBI.
AQP4 is a water-channel protein expressed strongly in the brain, predominantly in astrocyte foot processes surrounding capillaries [33]. Western blotting and immunohistochemistry results showed that EGCG significantly inhibited APQ4 expression after TBI. It has been reported that mice deficient in AQP4 have much better survival than wild-type mice in a model of brain edema caused by acute water intoxication and AQP4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke [34]. An underlying mechanism of EGCG on TBI may be that the inhibition of AQP4 expression leads to reducing water content and alleviating cerebral edema. Numerous studies report that TBI induces secondary molecular changes that lead to reactive astrogliosis, which is characterized by rapid synthesis of GFAP [35]. Our data indicated that EGCG decreased GFAP expression in brain tissue after TBI. These findings highlight an important action of EGCG targeting astrocytes in TBI.
ROS generation and oxidative stress has been implicated in contributing significantly to neuronal cell death and functional impairments following TBI [7]. Some enzymes such as NADPH oxidases and inducible nitric oxide synthase (iNOS) are up-regulated in brain tissues and produce free radicals which cause oxidative damage of DNA, proteins and lipid [36]. EGCG, as an anti-oxidant, increased SOD activity and decreased MDA activity following TBI (Fig. 5). As a major source of superoxide, NADPH oxidase is a membrane protein composed of several subunits including a Rho guanosine triphosphatase (GTPase) and five "phox" units. Following TBI, NADPH oxidase activation is dependent upon forming an active complex with several phox subunits (p47phox, p67phox, p40phox) and activated Rac1, which translocate to the membrane [37]. Our data showed that rac1 and p47phox were translocated to plasma membrane following TBI, and subsequently the EGCG inhibited p47phox translocation but not rac1. NADPH oxidase activity contributed significantly to the pathology of TBI via mediation of oxidative stress damage, microglial activation and AD protein induction in the brain following TBI [38]. A previous study indicated that inhibition of NADPH oxidase prevents TBI-induced ROS production and decrease BBB disruption, neuronal death and microglial activation [7]. Our data suggested that EGCG inhibited NADPH oxidase activation through inhibition of p47phox translocation. Taking the above findings together, it may be proposed that EGCG augments the cellular anti-oxidative system via inactivation of NADPH oxidase, resulting in decreased oxidative stress.
In conclusion, our results indicated that administration of EGCG immediately after TBI suppressed edema formation and exhibited neuroprotection against TBI-induced oxidative stress characterized by the severe disturbance of redox balance, at least in part, through the inhibition of NAPDH oxidase activation. Our findings further suggested that EGCG could be incorporated into novel therapeutic and preventive strategies for traumatic brain injury.
 XML Download
XML Download