

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
AM
acetoxymethylester
ADP
adenosine diphosphate
ATP
adenosine triphosphate
Carboxy-SNARF-1-AM
carboxy-seminaphtorhodafluor-1-acetoxymethylester
CCD
charge-coupled device
CPX
Ca2+-H+ exchanger
EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid
FCCP
Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
HEPES
N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
Kd
dissociation constant
mCU
mitochondrial Ca2+ uniporter
mNCX
mitochondrial Na+-Ca2+ exchanger
MPTP
mitochondrial permeability transition pore
NADH
Nicotinamide adenine dinucleotide
PMTs
photomultiplier tubes
RaM
rapid mode of Ca2+ uptake
RyR
ryanodine receptor channel
S.E.
Standard error
SR
sarcoplasmic reticular
TMRE
Tetramethylrhodamine ethyl ester
Ψm
mitochondrial membrane potential
INTRODUCTION
Ca2+ is an essential element involved in initiating and controlling the excitation-contraction coupling process in cardiac myocytes. The predominant Ca2+ source is trans-sarcolemmal Ca2+ influx and SR Ca2+ release [123]. For many years, mitochondria were simply regarded as an ATP-generating factory. However, several recent reports showed that mitochondria participate in many other important processes, such as apoptosis, ischemic-reperfusion injury, aging, and other diseases [45678]. All of those phenomena are related to Ca2+ [45678].
Several mechanisms of mitochondrial Ca2+ regulation in cardiac myocytes were reported, including involvement of the mCU [4679], RaM [1011], the RyR [1213], the mNCX [14151617], and the CPX [1819]. To date, their dynamics and quantitative roles in cardiac myocyte mitochondrial Ca2+ regulation were unclear. Further, their exact working mechanism, molecular identity, and regulatory factors were unknown. The role of mitochondria in cytosolic Ca2+ regulation in cardiac myocytes was controversial as well [467920]. In order to study these issues, quantitative and real-time measurements of [Ca2+]m were required.
The [Ca2+]m range was reportedly 0.08 to 20 µM [21222324]. The most frequently used fluoroprobe to determine this range was rhod-2. Rhod-2-AM has a positive charge, and therefore it may preferentially load into mitochondria because of the large negative potential of the mitochondrial matrix. However, rhod-2 has limitations regarding quantitative mitochondrial Ca2+ measurement because it is not a ratiometric dye. In addition, rhod-2 can measure only up to several µM of Ca2+ because its dissociation constant (Kd) ranged from 0.57 to 0.80 µM [122526].
Fura-2 analogs are widely used for quantitative measurements of [Ca2+]. The Kd value of Fura-2-FF was reportedly 18~35 µM in calibrating buffers [2728293031] and 6~13 µM in cells [2732]. However, this dye has potential usage problems due to NADH contamination and dye loading into mitochondria. Specifically, NADH contamination is a serious problem because most NADH is located in the mitochondria and because [NADH] change is related to mitochondrial membrane potential (Ψm), in turn, [Ca2+]m change [33343536]. These problems have been ignored and no methods exist to avoid or overcome them.
In this report, we developed a novel online NADH correction method to measure [Ca2+]m quantitatively, as well as a method to simultaneously measure multiple parameters, including NADH, [Ca2+]m, and pH/Ψm.
Go to : 
METHODS
Cell preparation
Sprague-Dawley (SD) rats (8~10 weeks old) were anesthetized with a mixture of ketamine hydrochloride (90 mg/kg) and Rompun® (10 mg/kg). We injected the anesthetics intraperitoneally. The rat chest cavity was opened, and the heart was excised and mounted on a Langendorff-type apparatus. The heart was sufficiently perfused with Tyrode's solution (mmol/L: NaCl, 133.5; KCl, 5.4; HEPES, 5; taurine, 20; MgCl2, 3; CaCl2, 0.75; and glucose, 5.5; pH adjusted to 7.4 with NaOH) to remove all residual blood in the heart. Subsequently, the heart was perfused with Ca2+-free Tyrode's solution (mmol/L: NaCl, 133.5; KCl, 5.4; HEPES, 5; taurine, 20; MgCl2 3; EGTA 0.5; glucose, 5.5; pH adjusted to 7.4 with NaOH), and then with collagenase (Worthington Biochemical Co., NJ, USA) containing Tyrode's solution (mmol/L: NaCl, 133.5; KCl, 5.4; HEPES, 5; taurine, 20; MgCl2, 3; CaCl2, 0.15; and glucose, 5.5; collagenase, 0.8 mg/mL; pronase, 0.04 mg/mL; pH adjusted to 7.4 with NaOH) for 15 minutes. Single cardiac myocytes were dispersed by gentle agitation of the digested heart in a high K+-low Cl- solution and stored in cell culture medium (Dulbecco's modified Eagle's medium; Sigma, St. Louis, USA) at room temperature (24~26℃) for later use. The high K+-low Cl- solution was comprised of the following, in mmol/L: KCl, 25; K-glutamate, 70; KH2PO4, 10; taurine, 10; glucose, 11; EGTA, 0.5; and HEPES, 10; pH adjusted to 7.2 with KOH. All experimental protocols were approved by our institutional animal care and use committee.
Solutions and reagents
Fura-2-FF was purchased from TEFLabs (TX, USA). The TMRE and carboxy-SNARF-1-AM were obtained from Invitrogen (Seoul, Korea).
Fluoroprobe loading procedure
Ventricular myocytes were incubated in Fura-2-FF-AM (8 µM) solution for 60 min at 4℃, and then at 37℃ for 30 min. Next, ventricular myocytes were incubated for 60 min at 37℃ with a dye-free solution to sufficiently hydrolyze the AM ester dye. Finally, the ventricular myocytes were transferred into and incubated in fresh medium. If carboxy-SNARF-1 was needed for the study, the dye (2 µM) was added into the second incubation step. To remove cytosolic compartments and to visualize the mitochondrial fluorescence, single isolated cardiomyocytes were perfused with saponin (0.1 mg/mL) for 60 s in the perfusing bath which is mounted on the inverted microscope (Fig. 1). All experiments were performed at 37℃.
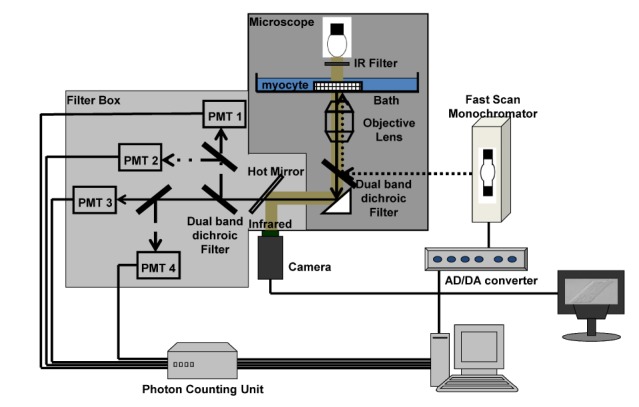 | Fig. 1A microfluorometry system for multiparameric measurement. The diagram shows the instrumentation used to measure NADH (emission, 450 nm; excitation, 361 nm), Fura-2-FF (emission, 500 nm; excitation, 353 and 400 nm), and TMRE (emission, 590 nm; excitation, 530 nm) or carboxy-SNARF-1 (emission, 590 and 640nm; excitation, 539 nm). The light from a Xenon arc lamp excited the fluoroprobes in a myocyte, and then the emission passed through a dichroic mirror and band-pass filters to measure the intensity using photon-counting devices. Near infrared from a microscope was used as a light source to visualize the myocytes during experiments using a CCD camera and monitor.
|
Spectrofluorometric measurements with a multiparametric measurement system
A fast monochromator (Polychrome II; Till Photonics, Inc., Germany) was used as an excitation light source. A fused quartz light guide was used to divert the excitation light to an inverted microscope (TE-300; Nikon, Tokyo, Japan). An oil immersion lens (40x, NA 1.3) was used. A near infrared filter (Chroma Technology Corp., Bellows Falls, VT, USA) was used between the microscope illuminator and specimen to monitor the object field with a charge-coupled device (CCD) camera (FTM1800NH/HGI; Philips, Salt Lake, USA). The image was captured with a BT878-based TV capture board, and the object field or cell area, as a pixel unit, was measured with custom-made software. The object field area was set with a field diaphragm (Nikon, Tokyo, Japan). Four photomultiplier tubes (PMTs) were used to detect emission wavelengths, and each had band-pass filters (450, 500, 590, and 640 nm). Dichroic mirrors and band-pass filters were purchased from Chroma (Brattleboro VT, USA). A photon counting method was applied with the combination of a photomultiplier tube (R2949; Hamamatsu, Hamamatsu Japan), photon counter unit (C3866; Hamamatsu, Hamamatsu, Japan), and high-speed counter (NI 6602; National Instruments, Austin, USA). Custom-made driving software was used to control and sample data. A diagram of the whole system is shown in Fig. 1.
Go to :
RESULTS
Development of a NADH correction method
NADH-Fura-2 signal interference
Simultaneous measurement of Fura-2-FF and NADH was technically difficult because each fluoroprobe produced signal interference. Fig. 2 shows this interference. Emission wavelengths of 450 and 500 nm were selected for NADH and Fura-2-FF, respectively. The excitation spectrums of both fluoroprobes overlapped. In addition, each fluoroprobe could generate emission at both 450 and 500 nm. The light intensity at each emission, therefore, contained signals from both fluoroprobes. This interference had to be corrected to obtain an exact measurement of the emission signal, specifically the quantitative measurement of Ca2+ with Fura-2-FF.
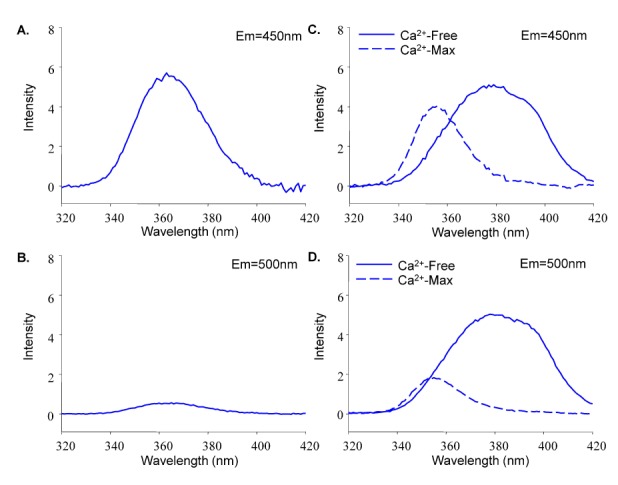 | Fig. 2The excitation spectrum of NADH-Fura-2-FF. The spectrum shows the interference between NADH and Fura-2-FF at each emission (450 and 500 nm). (A) The NADH excitation spectrum at 450-nm emission. (B) The NADH excitation spectrum at 500-nm emission. (C) The Fura-2-FF excitation spectrum at 450-nm emission under Ca2+-free and 1 mM Ca2+ conditions. (D) The Fura-2-FF excitation spectrum at 500-nm emission under Ca2+-free and 1 mM Ca2+ conditions.
|
Correction principles
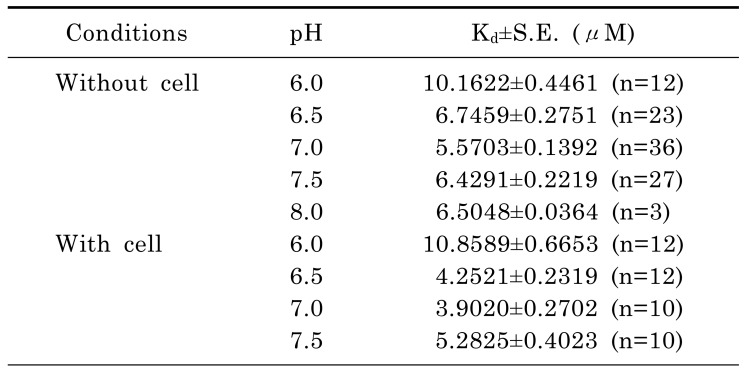
The excitation wavelength and the concentration of NADH can affect the emission intensity but not the emission spectral curve. Changes in [Ca2+] did not affect the emission and excitation spectrums of NADH (data not shown). Thus, the 450 and 500 nm ratio of NADH was constant at any excitation wavelength. The same principle can be applied to Fura-2-FF if NADH or [Ca2+] does not affect the emission and excitation spectrums of Fura-2-FF. However, the Fura-2-FF emission spectrum was shifted to the left by Ca2+ (Fig. 3B). Therefore, only isosbestic excitation could generate a Ca2+-independent emission spectrum. The isosbestic point was different for each emission wavelength (i.e., 450 and 500 nm). To minimize NADH contamination during isosbestic point acquisition, 10 µM FCCP and 10 µM ADP were added in the absence of mitochondrial substrates. The residual emission intensity was measured, and the relationship between the intensity and the cell area was obtained for correction of subsequent experiments. After myocytes were loaded with Fura-2-FF, the cell area was measured in each cardiac myocyte to correct the residual signals. The excitation spectrums at 450 and 500 nm emission were obtained by changing [Ca2+] from 0 to 10 mM under FCCP-free and mitochondrial substrate-free conditions (Fig. 4A and B). The excitation spectrum of the Ca2+-bound state was subtracted from the excitation spectrum of Ca2+-free conditions. Those subtracted curves are shown in Fig. 4C and D. To obtain an isosbestic point, we chose the wavelength showing the minimum standard deviation value (Fig. 4E and F). The isosbestic points were 361 at 450-nm emissions and 353 at 500-nm emissions, respectively. Using isosbestic excitation, the following equations were valid:
where Fx,y is the measured emission intensity at y nm by x nm excitation, Fx,y,NADH represents the pure NADH-dependent emission intensity, and Fx,y,fura represents the pure Fura-2-FF dependent emission intensity. In addition, the Rf, RN1, and RN2 values must be constant because of the consistent shape of the emission spectrum:
With these constants, equations (1), (2), and (3) can be changed as follows:
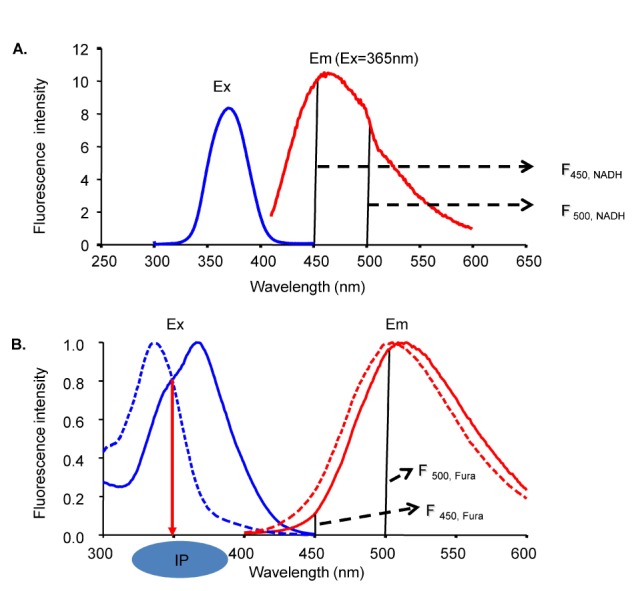 | Fig. 3NADH and Fura-2 excitation and emission spectra. (A) The NADH excitation spectrum was obtained at 465-nm emission and the emission spectrum was obtained at 365-nm excitation. (B) The Fura-2 excitation spectrum was obtained at 500-nm emission and the emission spectrum was obtained at 340-nm excitation. The emission spectrum was shifted to the left by an increase in Ca2+. Ex is excitation. Em is emission.
|
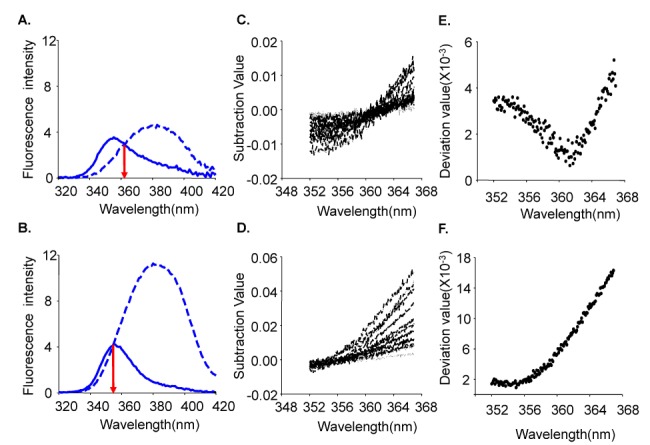 | Fig. 4Identification of isosbestic points. (A) The isosbestic point at 450-nm emission (red arrow). Ca2+ in the non-bound state (- -) and Ca2+ in the bound state (-) (B) The isosbestic point at 500-nm emission for Ca2+ in the non-bound state (- -) and Ca2+ in the bound state (-) (C) The graph shows subtracted data. Ca2+ in the non-bound state was subtracted from that in the bound state at 450 nm. (D) The graph shows subtracted data. Ca2+ in the non-bound state was subtracted from that in the bound state at 500 nm. (E) Standard deviation data from graph C. (F) Standard deviation data from graph D.
|
If the constants, Rf, RN1, and RN2, were known, the pure NADH and Fura-2-FF signals could be obtained by solving the above equations. The Rf was calculated with the fluorescence intensity observed in Fig. 4A and B. RN1 and RN2 were obtained in myocytes that were not loaded with Fura-2-FF. The NADH signal was changed according to the supply of mitochondrial substrates: malate, pyruvate, or malate plus pyruvate (Fig. 5A and B). The linear relationships of F400,500,NADH vs. F361,450,NADH and F353,500,NADH vs. F361,450,NADH are shown in Fig. 5C, and each slope indicates RN1 and RN2. The equations of each pure signal values are as follows:
Rfura is the ratio value of Fura-2-FF that was used to calculate the free calcium concentration.
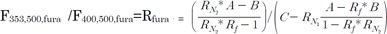
 | Fig. 5Measurement of RN factors. (A) The NADH signal in Fura-2-FF-free myocytes at 361-nm excitation and 450-nm emission was measured in the presence of various mitochondrial substrates. (B) F400,500 (●) and F353,500 (-) were measured simultaneously to monitor the NADH contribution to the Fura-2-FF signal. (C) The graph shows the relationships between F361,450,NADH and F353,500,NADH (●) and between F361,450,NADH and F400,500,NADH (○). Linear relationships are evident in the graph.
|
Background subtraction
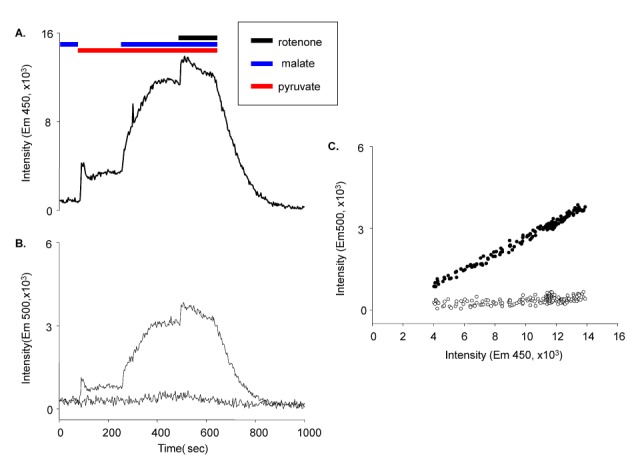
The excitation light intensity used to induce fluorescence was much stronger than the fluorescence emission. Although the selected excitation wavelength was far different from the emission wavelength and was blocked using a band-pass filter, the stray light from excitation was still present. The stray light was reflected at the boundary between the solution and the cover slip and diffracted in the cells due to cellular components. Cells may also produce unwanted autofluorescence. Those signal artifacts were comparable to the real fluorescence emission from the dye; thus, correction was necessary. First, the reflected signal at the cover slip boundary was measured. Then, the background signal due to cellular autofluorescence was measured in the presence of FCCP and in the absence of mitochondrial substrates. The cell-dependent background signal was linearly related to the cell area; these relationships are shown in Fig. 6. Later, the dye-loaded cellular area was measured, and the autofluorescence calculated from the cellular area was corrected. Further, the cell-free window background was also measured and subtracted. The background correction was performed before the experiments began. We determined the relationship between the cellular area and cellular autofluorescence on each experimental day.
Calibration equation
The Ca2+ concentration could be calculated from the ratio value calculated from equation (11) using Grykiewicz's equation [37]. To convert the ratio into a concentration, the equation required four parameters: Kd, dissociation constant; Rmin, minimum ratio; Rmax, maximum ratio; and F380,max/F380,min, the ratio of maximum and minimum at 380 nm excitation. However, applying the equation was not easy because both Rmax and F380,max/F380,min require Ca2+-saturated conditions without dye-loss. Replicating that condition in real cells was a difficult and erroneous procedure. In mitochondria, dye-saturated conditions for Fura-2-FF without dye-loss were virtually impossible. We found that the Ca2+-bound form of Fura-2-FF was practically non-fluorescent at 400-nm excitation. The new equation using this property was developed and is as follows: The F400,500,max and F353,500,max are maximum values of the emitted signals at 500 nm with the excitation at 400 and 353 nm. Since F353,500,max was an isosbestic emission value and was constant, the above equation could be simplified further as follows: The details of the derivation of the above equations are shown in the Appendix.
The Kd of Fura-2-FF
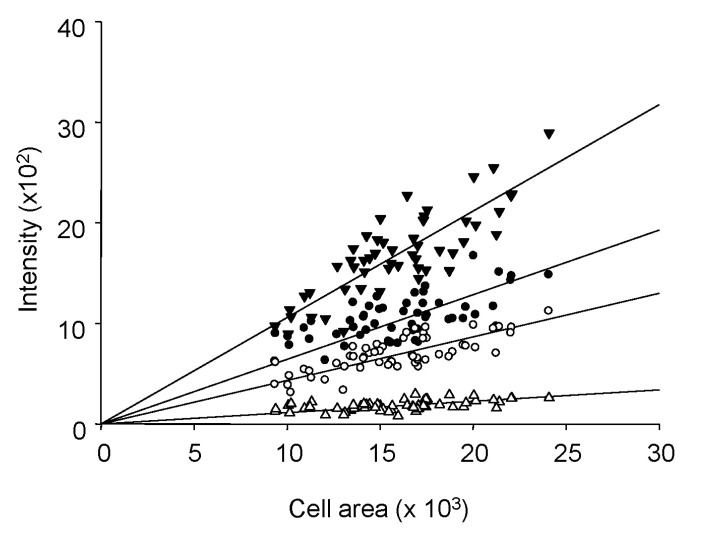
To apply the calibration equation, the Kd value had to be determined. If we knew the free [Ca2+] and the other parameters in the calibration equation, we could calculate Kd. To calculate the free Ca2+ concentration, WINMAXC32 version 2.50 (Chris Patton, Stanford University) was used. EGTA purity was double-checked using the pH and oxalate method [3839404142]. We obtained pH-dependent changes of Kd values in a cell-free solution. To simulate the experimental conditions, we measured pH-dependent Kd values with a dye-free cell in the window. A summary of the measured Kd values is shown in Table 1.
Mitochondrial pH changes by the change in [Ca2+]m
Using carboxy-SNARF loaded myocytes, mitochondrial pH changes were determined following Ca2+ changes (Fig. 7). The increase in [Ca2+]m did not affect mitochondrial pH. The mitochondrial pH was 7.504±0.047 (mean±standard error [S.E.], n=13). The Kd value of Fura-2-FF at pH 7.5 was 5.28 µM; this value was used for later Ca2+ calculations.
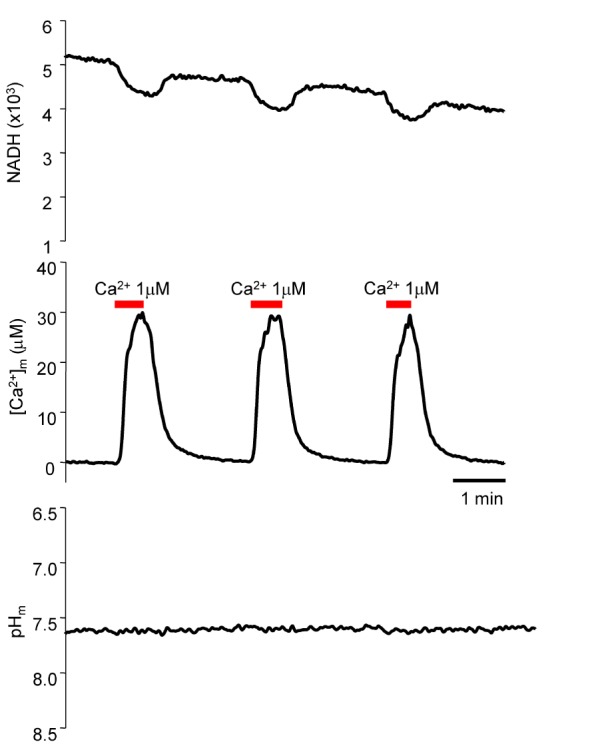 | Fig. 7Simultaneous measurement of NADH, [Ca2+]m, and pH. Using carboxy-SNARF- and Fura-2-FF-loaded myocytes, NADH, [Ca2+]m, and pH was measured simultaneously. The mitochondrial pH changes were investigated following Ca2+ changes. The increase in [Ca2+]m did not affect mitochondrial pH. Mitochondrial pH was 7.504±0.047 (mean±S.E., n=13).
|
Mitochondrial Ca2+ changes due to correction
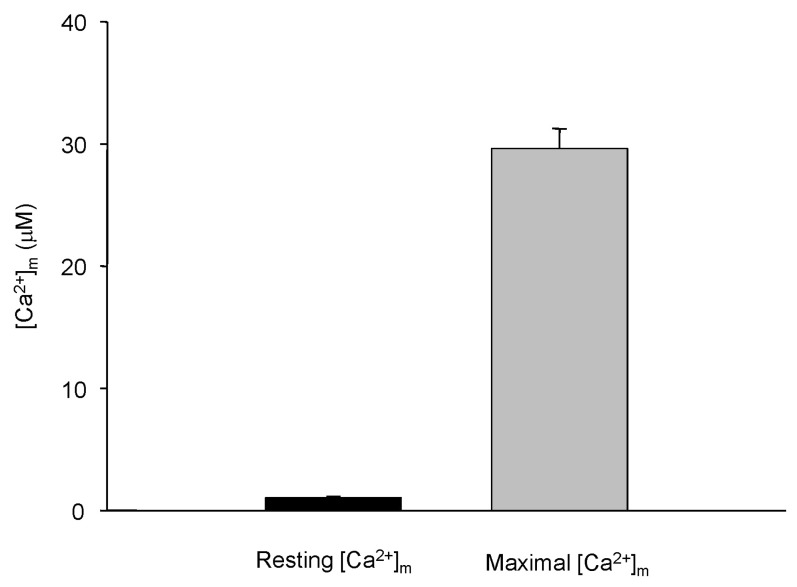
Fig. 8 shows the differences in Ca2+ concentrations before and after the correction. The results clearly showed the importance of correction for NADH and Fura-2-FF interference. The mitochondrial resting calcium concentration was 1.03±0.13 µM (mean±S.E., n=32) and the maximum [Ca2+]m at 1 µM cytosolic Ca2+ ([Ca2+]c) was 29.6±1.61 µM (mean±S.E., n=33) (Fig. 9).
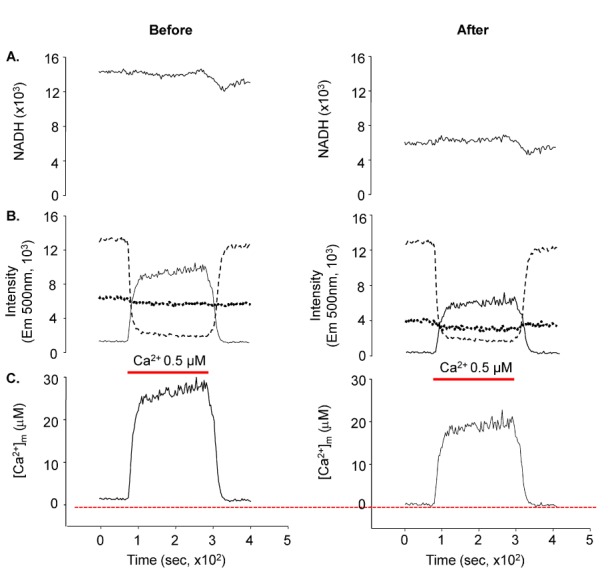 | Fig. 8Results of NADH and Fura-2-FF interference correction. The left panels show the signals prior to correction and the right panels show the signals after correction. NADH and the ratio of Fura-2-FF signals were changed following correction. The calculated [Ca2+]m was substantially decreased. (A) NADH signals at 450-nm emission. (B) Fura-2-FF signals at 500-nm emission. The figure shows F400,500 (- -), F353,500 (----), and the ratio of Fura-2-FF (-). (C) The mitochondrial calcium concentration calculated from the Fura-2-FF ratio.
|
Simultaneous measurement of NADH, [Ca2+]m, and Ψm
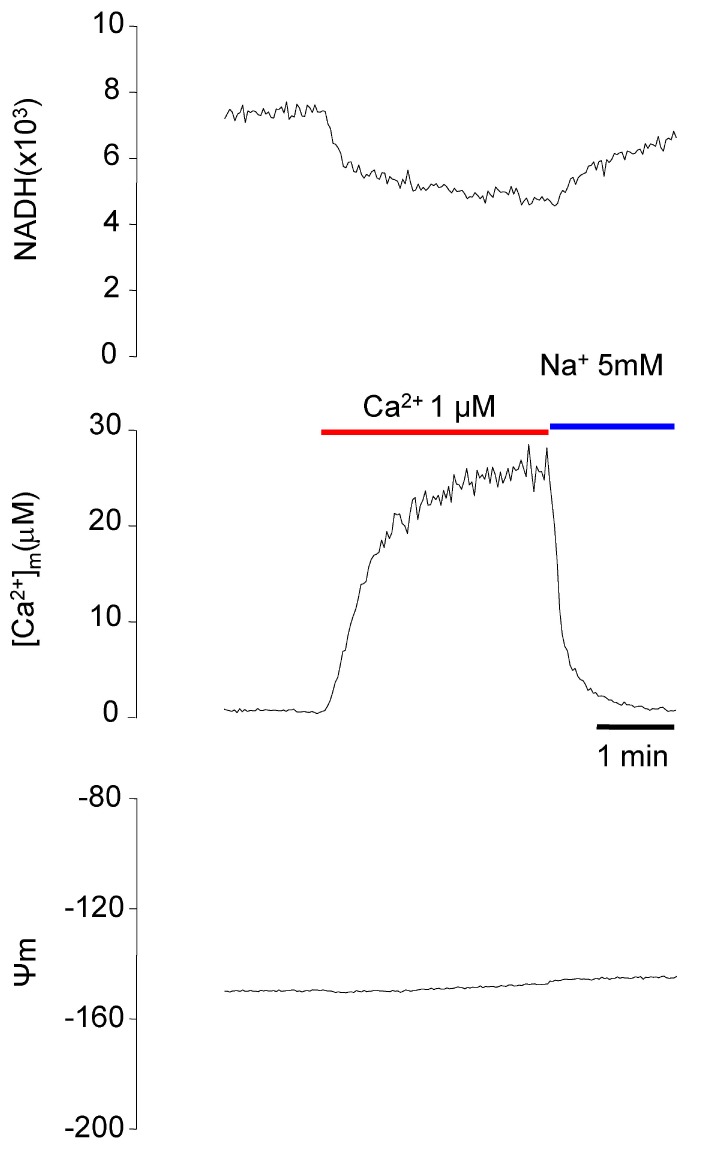
TMRE has a positive charge and is distributed in a membrane potential-dependent manner. If the concentration in each compartment is known, the membrane potential can be calculated using Nernst's equation. TMRE was perfused at 2 nM to record the changes in the TMRE signal in mitochondria. The resting Ψm was assumed to be -150 mV. Based on this, Ψm was calculated. NADH was decreased by the application of Ca2+, but the Ψm remained similar (Fig. 10).
Go to :
DISCUSSION
Interest in the ubiquitous role of Ca2+ in cells and cellular organelles is increasing. To study Ca2+ dynamics, the use of Ca2+ indicators has steadily increased. Mitochondria have now become a focus because of their role in many cellular processes, such as energy generation, apoptosis, ischemic-reperfusion injury in the heart, aging, etc. Ca2+ has a central role in these processes. However, quantitative and dynamic mitochondrial Ca2+ data are not yet available. For quantitative measurement of mitochondrial Ca2+, a ratiometric approach is essential. Ratiometric fluoroprobes are advantageous for quantitative measurement of [Ca2+] because dye loss due to leakage or photobleaching does not affect the calculation of [Ca2+]. These problems cannot be avoided when single excitation and single emission dyes are used to determine [Ca2+].
Fura-2 was first introduced in 1985 as a ratiometric fluorescent dye [37]; several analogs were subsequently developed. However, Fura-2 has several disadvantages when used to measure mitochondrial Ca2+. First, targeted dye loading into the mitochondria is not easy. Therefore, Fura-2 would likely be nonspecifically loaded into all organelles of the cell. One major Ca2+-related organelle is the SR. However, the total SR and mitochondria percent volumes are about 3.5% and 34~36%, respectively, in rat ventricular myocytes [4344]. Therefore, even though Ca2+ in SR may affect the results, the effect would be negligible. Second, cellular autofluorescence from NADH could contaminate the Fura-2 signal. More than 90% of cellular NADH is present in the mitochondria (data not shown). Excitation and emission spectrums of NADH and Fura-2 widely overlap. Importantly, Ca2+ changes in mitochondria could affect NADH, which, in turn, affects Fura-2 signals. Therefore, this complicated interference must be solved to quantitatively measure [Ca2+]m. This problem, however, has not been systematically investigated since Fura-2 was introduced. We were able to successfully correct for the interference between NADH and Fura-2 using the spectral characteristics of NADH and Fura-2. Fig. 8 shows the effect of NADH and Fura-2-FF interference. The NADH signal was considerably decreased after the correction (Fig. 8). The corrected Fura-2-FF signal resulted in lower [Ca2+]m. This interference implied that data from previous reports using Fura-2 analogs could be wrong, as the NADH signal could be different based on the correction or lack thereof. We would like to further investigate this possibility in the future.
Our calibration equation (Equation 12) has many advantages over Grynkiewicz's equation. First, it only required three calibration parameters: Kd, F340,max/F400,max, and Rmin. Second, the ratio value was directly linear to the Ca2+ concentration at a constant pH. Therefore, the rate of ratio change could be directly converted to the rate of Ca2+ change. Third, obtaining the calibration parameters, particularly F340,max/F400,max, is relatively error-free. In Grynkiewicz's equation, F380,max/F380,min is prone to error because it divides the largest number by the smallest number. In addition, to obtain F380,max/F380,min, we had to accomplish real free- and saturated-[Ca2+] conditions because F380 must reach the real maximum and minimum values, respectively. If not, F380,max/F380,min can easily be faulty. However, in our new equation, the F340,max/F400,max measurement error is much smaller because it is the ratio of the largest numbers at each excitation. Fourth, as shown in Equation 13, when the isosbestic excitation value was chosen instead of 340 nm in the new equation, only Rmin is required to convert the ratio into the Ca2+ concentration if the pH was unchanged and the Kd value at that pH is known. Finally, the calibration procedure is much simpler, as Rmin is the only parameter needed.
However, there are limitations associated with use of the equation. The new equation was generated based on the assumption that the emission of Ca2+-saturated Fura-2-FF was zero at 400-nm excitation. However, Ca2+-saturated Fura-2-FF actually produces a small emission. This causes deflection from the linearity between the ratio and Ca2+ concentration at very high Ca2+ concentrations. We that found our equation could be reasonably applied to [Ca2+]m concentrations up to 50-fold that of the Kd. This range is reasonably acceptable under experimental conditions. The new equation still does not solve the problems of incomplete hydrolysis of the AM form of the dye, dye compartmentalization, autofluorescence, etc. Dye loading problems are inevitable, but careful loading and washing may minimize these kinds of errors (see Ref. [45]). In the case of compartmentalization, mitochondria had the largest volume; therefore, this type of error could be ignored. We used a very careful loading procedure to minimize incomplete hydrolysis of the AM form; therefore, we believe that this error was also minimized.
The Kd value of Fura-2-FF is pH dependent; therefore, if the procedure could cause changes in pH, we need to consider the pH effect when calculating [Ca2+]m. Cytosolic Ca2+ application increased [Ca2+]m, but the mitochondrial pH did not change. Therefore, the constant Kd at pH 7.5 was used to calculate [Ca2+]m.
Our microfluorometry system could measure NADH, [Ca2+]m, and pHm simultaneously. In addition, the Ψm could be measured using TMRE because the excitation and the emission spectrum of TMRE overlapped with those of carboxy-SNARF-1. Therefore, we were able to successfully measure Ψm using TMRE (Fig. 10).
The resting [Ca2+]m was approximately 1.03 µM. A previous report showed that [Ca2+]m ranged from 0.08 to 0.17 µM [2324]. Our study revealed a higher value than that of previous reports (see Refs. [2324]). The change in [Ca2+]m plays a central role in the exercise-dependent increase in ATP by activating three mitochondrial matrix Ca2+-sensitive enzymes (pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and NAD-dependent isocitrate dehydrogenase) [464748495051]. Pyruvate and α-ketoglutarate dehydrogenase were almost fully activated at 1 µM because of the low Kd values, 0.77 and 0.28 µM, respectively [51]. The Kd value of NAD-dependent isocitrate dehydrogenase was 5.4 µM [51]. Therefore, the [Ca2+]m-dependent increase in ATP may be due to the activation of NAD-dependent isocitrate dehydrogenase.
The maximal attainable [Ca2+]m seemed to be limited. Our data showed that the maximal [Ca2+]m was about 30 µM at 1 µM cytosolic Ca2+. Even when we applied more than 1 µM, the maximal [Ca2+]m was not increased further (data not shown). The 30 µM concentration is far below the electrochemical equilibrium because the Ψm is about -150 mV and the concentration of the electrochemical equilibrium must be up to 105 times the cytosolic [Ca2+]. This limitation implied the strong possibility that other mechanisms exist to limit Ca2+ increase in mitochondria. This may be a Ca2+ efflux mechanism, an inhibitory regulator of Ca2+ influx, or the increase in Ca2+ buffer by a volume increase or an actual buffer increase. Until now, two mechanisms were suggested for mitochondrial Ca2+ efflux, mNCX and CPX. Ca2+ was applied under 0 mM Na+ conditions; therefore, mNCX could not be the efflux mechanism involved in limiting the increase in [Ca2+]m. CPX may change mitochondrial pH because it causes influx of H+ into mitochondria. However, mitochondrial pH was not changed during [Ca2+]m increase. The MCU, a major Ca2+ influx pathway, did not show inactivation [52]. The electrochemical gradient for Ca2+ influx was huge, and thus it would likely be difficult for CPX to counteract the Ca2+ influx to limit the [Ca2+]m increase. Another possibility is the opening of the MPTP. It was reported that [Ca2+]m overload could open the MPTP [535455]. However, MPTP is permeable to 1.5-kDa molecules [545556]; if the MPTP was opened, Fura-2-FF or NADH would be decreased. Further, if the MPTP was opened, the Ψm would be substantially depolarized. However, we did not observe these phenomena. Mitochondria are known to be a Ca2+ sink because of their relatively larger Ca2+ buffering capacity [57585960]. If the limitation was caused by the buffer, [Ca2+]m must continuously increase, even though the speed was slower; however, this was not observed. Ca2+ and Pi could form a precipitate [61], and free [Ca2+]m may not increase because of continuous precipitate formation. Another possibility is mitochondrial swelling. Ca2+ overload caused mitochondrial swelling, an indication of MPTP opening [535455]. However, actual swelling may occur without MPTP opening. Mitochondria reportedly had a Pi-containing particle [61]. Mitochondrial swelling and dissolution of those particles may increase mitochondrial buffering capacity to limit [Ca2+]m increase. The Pi effect on mitochondrial Ca2+ dynamics requires further study.
In conclusion, we successfully developed a method to solve NADH and Fura-2-FF interference. This method enables more accurate Ca2+ measurements. In particular, quantitative measurement of mitochondrial Ca2+ dynamics is now possible and many uncertainties surrounding mitochondrial Ca2+ dynamics can be answered in the future using our new method.
Go to :
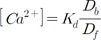
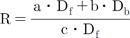
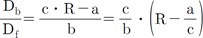
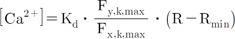
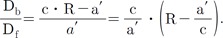
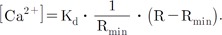
 XML Download
XML Download